

Аниридия врожденная: нередкая среди редких
Отсутствие радужной оболочки глаза или ее гипоплазия – так проявляется редкая врожденная патология – аниридия.
Глаз без радужки
Впервые это заболевание описал итальянский ученый Дж.Баррата в 1818 году.1 Самое видимое проявление патологии – отсутствие радужной оболочки или ее гипоплазия – дало и название описанному состоянию. Аниридия в переводе с греческого буквально означает «отсутствие радужной оболочки».
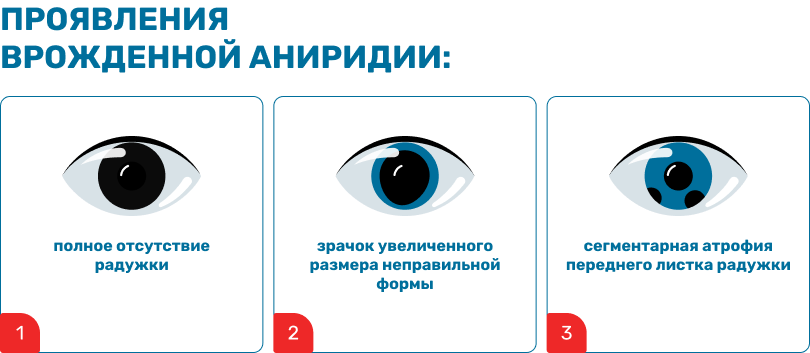
Многие годы потребовались офтальмологам для понимания проявлений этой аномалии, природы механизмов ее развития, патологических изменений, захватывающих роговицу, хрусталик, зрительный нерв и сетчатку.2 Но и сегодня у специалистов остается много нерешенных вопросов по этой нозологии. Внимание российских ученых эта аномалия органа зрения привлекла совсем недавно.

Это прогрессирующее заболевание, приводящее к значительному снижению зрения. Оно часто сопровождается персистирующей капиллярной мембраной, врожденной катарактой, эктопией хрусталика, глаукомой, паннусом роговицы, кератопатией и гипоплазией фовеа. Поражение многих внутриглазных структур должно предусматривать комплексный подход к диагностике и лечению врожденной аниридии. А понимание патогенетических механизмов течения болезни необходимо для прицельного лечения существующих осложнений и улучшения качества жизни пациентов.2
Врожденная аниридия (ВА) может быть изолированным нарушением, а может проявляться в составе различных синдромов (синдромов WAGR, Ригера, Гиллеспи, др.), поэтому требует не только офтальмологической коррекции, но и профессиональной помощи врачей других специальностей.
ВА зарегистрирована на сайте орфанных (редких) заболеваний Orphanet (ORPHA77) и с 2014 года официально включена Минздравом России в список редких заболеваний.2
1. Nelson L., Spaeth G., Nowinski T., Margo C. et al. Aniridia: A review. Surv. Ophthalmol. 1984, 28, p. 621—42.
2. Воскресенская А.А., Поздеева Н.А., Хлебникова О.В., Васильева Т.А., Зинченко Р.А. Клинические аспекты врожденной аниридии в России. Практическая медицина. Том 1, № 2 (87), 2015, с. 7—15.
3. Аниридия врожденная. Клинические рекомендации. https://cr.minzdrav.gov.ru/preview-cr/740_1?ysclid=m5yztw8h1o73408754 (Дата обращения: 15.01.2025.).
4. Gramer E., Reiter C., Gramer G. Glaucoma and frequency of ocular and general diseases in 30patients with aniridia: a clinical study. Eur. J. Ophthalmol. 2012, vol. 22, iss. 1, p. 104—110.
Проблемный ген

Аниридия наследуется по аутосомно-доминантному типу с почти полной пенетрантностью и различной экспрессивностью. В 90% случаев виновниками аномалии выступают мутации гена РАХ6 (отвечающего за строение и функционирование будущего глаза плода и контроль развития части центральной нервной системы), в том числе хромосомные перестройки региона 11p13 с вовлечением локуса PAX6 или в 5% удаленной цис-регуляторной области, контролирующей экспрессию гена PAX6.4-6 Формирование измененного участка происходит на 8—14 неделе эмбрионального развития.

Патогенез врожденной аниридии, клинические проявления, степень поражения различных структур глаза обусловлены генетической гетерогенностью и локусной (мутации различных генов), и аллельной (тип мутаций). Совокупность и сочетание генетических вариантов (генетический фон), которые приводят к снижению адаптации, играют важное значение в формировании клинической картины заболевания.8
Однако в случае аниридии специалистам пока не удается четко установить корреляционную связь между генотипом и фенотипическими проявлениями заболевания.9 Различия в фенотипе (выраженность дефекта радужки, степень кератопатии и др.) выявляются не только в рамках одной семьи со сходной мутацией в гене РАХ6, но также вариабельны у одного человека.
1. Васильева Т.А., Воскресенская А.А. Хлебникова О.В. и др. Дифференциальная диагностика наследственных форм врожденной аниридии с позиций современной генетики // Вестник РАМН. 72 (4), 2017, с. 233—241.
2. Valenzuela A., Cline R. Ocular and nonocular findings in patients with aniridia. Can. J. Ophthalmol. 2004, 39, p. 632—638.
3. Angmo D., Jha B., Panda A. Congenital aniridia. Journal of Current Glaucoma Practice. 2011, 5 (2), p. 1—13.
4. Gronskov K., Olsen J., Sand A. et al. Population-based risk estimates of Wilms tumor in sporadic aniridia. A comprehensive mutation screening procedure of PAX6 identifies 80% of mutations in aniridia. Hum. Genet. 2001, 109, p. 11—18.
5. Crolla J.A., van Heyningen V. Frequent chromosome aberrations revealed by molecular cytogeneticstudies in patients with aniridia. Am. J. Hum. Genet. 2002, vol. 71, iss. 5, p. 1138—1149.
6. Robinson D., Howarth R., Williamson K., van Heyningen V. et al. Genetic analysis of chromosome 11p13 and the PAX6 gene in a series of 125 cases referred with aniridia. Am. J. Med Genet A. 2008, 146A, p. 558—569.
7. Hingorani M., Moore A. Aniridia. GeneReviews (R) / Pagon R.A. et al. Seattle (WA), 1993.,4 Kokotas H., Petersen M. B. Clinical and molecular aspects of aniridia. Clin. Genet. 2010, vol. 77, iss. 5, p. 409—420.
8. Käsmann-Kellner B., Seitz B. Aniridia syndrome: clinical findings, problematic courses andsuggestions for optimization of care («aniridia guide»). Ophthalmologe. 2014, vol. 111, iss. 12, p. 1145—1156. PMID: 25475188.
9. Calvao-Pires P., Santos-Silva R., Falcao-Reis F., RochaSousa A. Congenital aniridia: clinic, genetics, therapeutics and prognosis. International Scholarly Research Notices. 2014, article ID 305350. http//dx.doi.org/10.1155/2014/305350.
Трудности классификации
Единой классификации врожденной аниридии нет. Предложенная в 1996 году Чарчиль А. систематизация с позиций этиопатогенеза и клинического портрета оказалась очень трудоемкой и не слишком приемлемой в рутинной практике. В последующем она претерпела значительные изменения, и, исходя из этиопатогенеза, клинической картины.

Тем не менее до сих пор не установлены конкретные взаимосвязи типов мутации гена PAX6 (хромосомной делеции) и особенностей клинической картины ВА.8
Есть и другие подходы к классификации ВА. Так, при врожденной аниридии степень презентации радужки значительно варьирует: от рудиментарных остатков шириной не более 0,5–1,0 мм до колобомоподобных дефектов и небольшой гипоплазии. Поэтому ряд специалистов предлагают использование понятий «полная аниридия», «частичная аниридия», «гипоплазия радужки», способных отразить степень потери радужной ткани.9-11
1. Practical paediatric ophthalmology. / Taylor D., Hoyt C. S. Oxford England. Cambridge. Mass.,USA: Blackwell Science, 1997. 232 p.
2. Kikuta H., Laplante M., Navratilova P., Komisarczuk A. Z., Engstrom P. G., Fredman D. et al. Genomic regulatory blocks encompass multiple neighboring genes and maintain conserved synteny in vertebrates. Genome Res. 2007, vol. 17, iss. 5, p. 545—555.
3. Gould D.В., John S.W. Anterior segment dysgenesis and the developmental glaucomas arecomplex traits. Hum. Mol Genet. 2002, vol. 11, iss. 10, p. 1185—1193.
4. Robinson D.O., Howarth R.J., Williamson K.A., van Heyningen V. et al. Genetic analysis of chromosome 11p13 and the PAX6 gene in a series of 125 cases referred withaniridia. Am. J. Med Genet A. 2008. vol. 146A, iss. 5, p. 558—569.
6. Dansault A., David G., Schwartz C., Jaliffa C., Vieira V., de la Houssaye G. et al. Three new PAX6 mutations including one causing an unusual ophthalmic phenotype associated with neurodevelopmental abnormalities. Mol Vis. 2007. vol. 13, p. 511—523.
7. https://omim.org/ (Дата обращения: 16.01.2025).
8. Васильева Т.А., Воскресенская А.А., Хлебникова О.В. и др. Дифференциальная диагностика наследственных форм врожденной аниридии с позиций современной генетики // Вестник РАМН. 72 (4), 2017, с. 233—241.
9. Аниридия врожденная. Клинические рекомендации. https://cr.minzdrav.gov.ru/preview-cr/740_1?ysclid=m5yztw8h1o73408754 (Дата обращения: 15.01.2025).
10. Hingorani M., Williamson K., Moore A., van Heyningen V. Detailed ophthalmologic evaluation of 43 individuals with PAX6 mutations. Invest Ophthalmol Vis Sci. 2009, 50, p. 2581—2590.
11. Hewitt A., Kearns L., Jamieson R., Williamson K. et al. PAX6 mutations may associated with high myopia. Ophthalmic Genet. 2007, 28 (3), p. 179—82.
Редкая патология
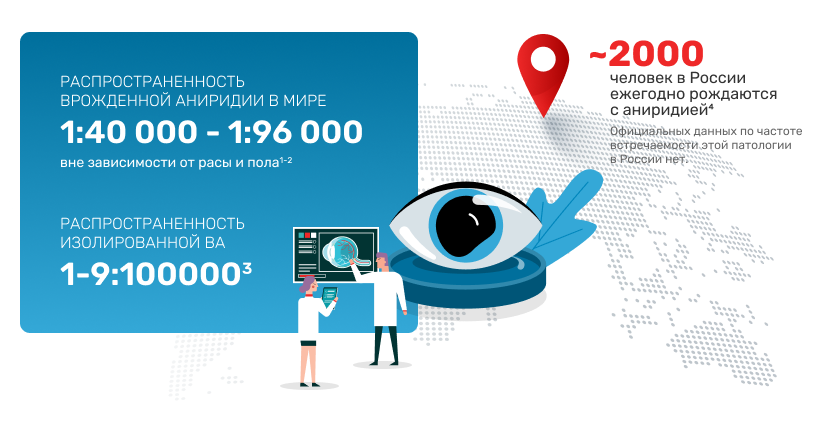
1. Nelson L., Spaeth G., Nowinski T., Margo C. et al. Aniridia: A review Surv Ophthalmol. 1984, 28, p. 621—642.
2. Gronskov K., Olsen J., Sand A. et al. Population-based risk estimates of Wilms tumor in sporadic aniridia. A comprehensive mutation screening procedure of PAX6 identifies 80% of mutations in aniridia. Hum Genet. 2001, 109, p. 11—18.
3. URL: https://www.orpha.net/ (дата обращения: 15.01.2025).
4. http://oftalmic.ru/b_aniridia.php?ysclid=m60sarisbh251242297 (Дата обращения: 14.01.2025).
Клиническая картина
Как правило у пациента с ВА и/или его родителей специфические жалобы отсутствуют. Патология выявляется педиатрами при физикальном обследовании. При частичной аниридии, гипоплазии радужки, то есть при слабой степени вовлечения в патологический процесс радужной оболочки, размер зрачка может быть нормальным.


Так, в анализе клинической картины врожденной аниридии у 57 пациентов (114 глаз), проведенном нашими коллегами, наблюдались следующие глазные проявления: при рождении чаще всего фиксируются дефекты радужной оболочки и гипоплазия фовеа, определяющие низкие зрительные функции ребенка и появление нистагма, в более поздние возрастные сроки чаще всего обнаруживаются катаракта, глаукома и патология роговицы (см.табл.).3

По данным разных авторов, в большинстве случаев острота зрения у пациентов с аниридией колеблется в пределах 0,1—0,2.4-5 Часто заболевание сопровождают аномалии рефракции: в более чем половине случаев (53,4%) это миопия, причем в 17% – близорукость высокой степени. Механизм этого наследования до конца неясен.6

1. Аниридия врожденная. Клинические рекомендации. https://cr.minzdrav.gov.ru/preview-cr/740_1?ysclid=m5yztw8h1o73408754 (Дата обращения: 15.01.2025).
2. Käsmann-Kellner B., Seitz B. Aniridia syndrome: clinical findings, problematic courses andsuggestions for optimization of care («aniridia guide»). Ophthalmologe. 2014. vol. 111, iss. 12, p. 1145—1156. PMID: 25475188.
3. Воскресенская А.А., Поздеева Н.А., Хлебникова О.В., Васильева Т.А., Зинченко Р.А. Клинические аспекты врожденной аниридии в России // Практическая медицина. Том 1, № 2 (87), 2015, с. 7—15.
4. Park S., Park Y., Lee M., Kim M. Clinical features of Korean patients with congenital aniridia. Korean J. Ophthalmol. 2010, 24 (5), p. 291—296.
5. Hingorani M., Hanson I., van Heyningen V. Aniridia. Eur J. Hum. Genet. 2012, 20, p. 1011—1017.
6. Hewitt A., Kearns L., Jamieson R., Williamson K. et al. PAX6 mutations may associated with high myopia. Ophthalmic Genet. 2007, 28 (3), p. 179—182.
7. Nelson L., Spaeth G., Nowinski T., Margo C. et al. Aniridia: A review. Surv Ophthalmol. 1984, 28, p. 621—642.
8. Brauner S., Walton D., Chen T. Aniridia. Int Ophthalmol Clin. 2008, 48, p. 79—85.
9. Grant W., Walton D. Progressive changes in the angle in congenital aniridia, with development of glaucoma. Am. J. Ophthalmol. 1974, 78, p. 842—847.
10. Grant W., Walton D. Progressive changes in the angle in congenital aniridia, with development of glaucoma. Trans Am. Ophthalmol Soc. 1974, 72, p. 207—228.
11. Hingorani M. Detailed ophthalmologic evaluation of 43 individuals / М. Hingorani, К. А. Williamson, А. Т. Moore et al. // Invest. Ophthalmol. Vis. Sci. 2009, vol. 50, iss. 6, p. 2581—2590.
Диагноз: установить в ранние сроки
При подозрении на наличие врожденной аниридии участковые педиатры, врачи общей практики направляют ребенка на консультацию к врачу-офтальмологу. Специалист оценивает состояние глаза, проводит обследование, включающее методы инструментальных исследований для уточнения диагноза. Если требуется проведение диагностических процедур с применением анестезии, дети направляются в детское офтальмологическое отделение стационара.
В случаях семейной ВА диагностических трудностей выявления аномалии не возникает. Врачам и родителям известно, что риск рождения ребенка с заболеванием – 50%. При спорадической ВА установление диагноза может быть затруднено и может затянуться на 7—9 месяцев.1

В полное офтальмологическое обследование входят (см.табл.)1-8:

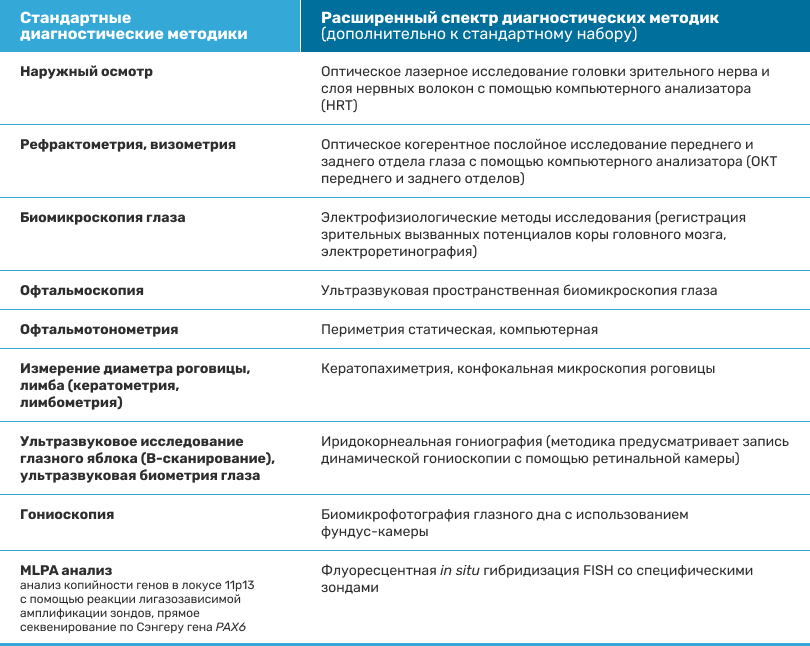
Таблица. Методы обследования1
P.S. Рекомендовано медико-генетическое обследование всех членов семьи пациента с ВА с учетом клинического полиморфизма (см.рис.).1
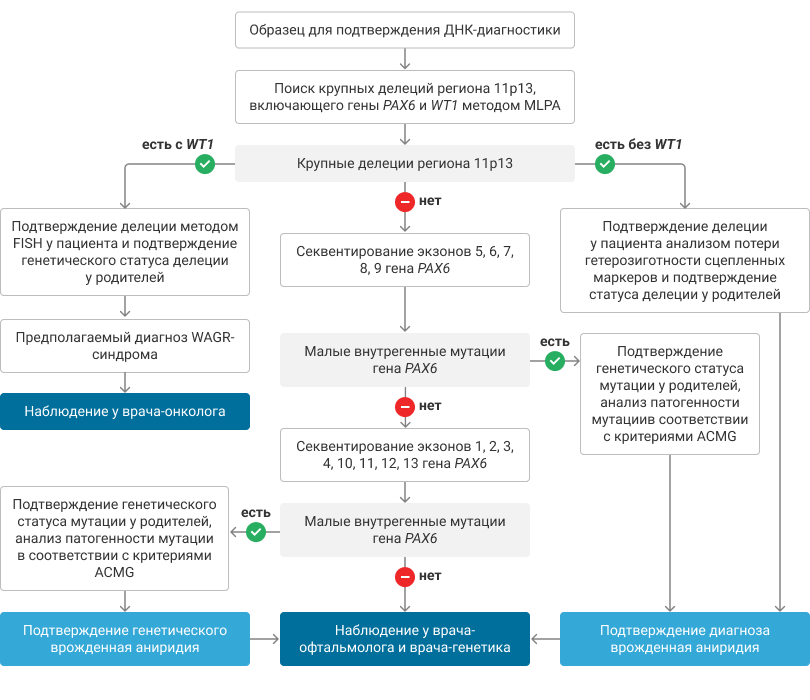
Рисунок. Молекулярно-генетическая диагностика врожденной аниридии. Алгоритмы действий врача.1
Особенности ВА показывают, что в раннем детском возрасте важно выполнение генетического обследования для выявления синдромальной аниридии – для своевременного обнаружения жизнеугрожающей патологии (WAGR-синдром и др.) и заболеваний, связанных со значительным снижением зрения (аномалия Петерса, синдром Ригера и пр.).1,9
Все дети с ВА должны быть обследованы педиатром, невропатологом, кардиологом, генетиком и при необходимости другими специалистами на предмет наличия соматической патологии.1,2
1. Аниридия врожденная. Клинические рекомендации. https://cr.minzdrav.gov.ru/preview-cr/740_1?ysclid=m5yztw8h1o73408754 (Дата обращения: 15.01.2025).
2. Воскресенская А.А., Поздеева Н.А., Хлебникова О.В., Васильева Т.А., Зинченко Р.А. Клинические аспекты врожденной аниридии в России // Практическая медицина. Том 1, № 2 (87), 2015, с. 7—15.
3. Chang J.W., Kim J.H., Kim S.J., Yu Y.S. Congenital aniridia: long-term clinical course, visual outcome, and prognostic factors. Korean J Ophthalmol. 2014, 28 (6), p. 479—485.
4. Lee H., Khan R., O’Keefe M. Aniridia: current pathology and management. Acta Ophthalmol. 2008, vol. 86, p. 708—715.
5. Singh B., Mohamed A., Chaurasia S., Ramappa M., Mandal A.K. et al. Sangwan VS Clinical manifestations of congenital aniridia. J. Pediatr. Ophthalmol Strabismus. 2014, 51 (1), p. 59—62.
6. Samant M., Chauhan B.K., Lathrop K.L., Nischal K.K. Congenital aniridia: etiology, manifestations and management. Expert Review of Ophthalmology. 2016, 11 (2), p. 135—144, DOI: 10.1586/17469899.2016.1152182.
7. McCulley T.J., Mayer K. et al. Aniridia and optic nerve hypoplasia. Eye. 2005, 19 (7), p. 762—764.
8. Mayer K.L., Nordlund M.L. et al. Keratopathy in congenital aniridia. Ocul Surf. 2003, 1 (2), p. 74—79.
9. Марахонов А.В., Васильева Т.А., Воскресенская А.А., Кадышев В.В. и соавт. Опыт применения медицинской технологии диагностики врожденной аниридии в ФГБНУ «МГНЦ» // Медицинская генетика. Т. 16, № 11, 2017, с. 23—26.
Ген РАХ6: союз офтальмолога и невролога
Ген РАХ6, с мутациями которого связана врожденная аниридия, отвечает не только за строение и функционирование будущего глаза плода, но также имеет отношение и к контролю за развитием части центральной нервной системы. Поэтому его мутации могут приводить как к необратимым аномалиям в структуре глаза, так и к дефектности нервной системы.

При ВА нистагм чаще горизонтальный, высокоамплитудный, возможно присутствие ротаторного компонента. По мере взросления ребенка нистагм уменьшается – снижается амплитуда колебаний и частота его проявлений. Но психоэмоциональная зависимость сохраняется.1

Рисунок. Неврологические особенности при аниридии1
Помимо нистагма у пациентов с аниридией может наблюдаться и другая неврологическая симптоматика (см.рис.):
- светобоязнь– у взрослых солнечный свет, дополнительное освещение провоцируют зажмуривание глаз, слезотечение, наклон головы, защиту глаз с рукой; негативная реакция на свет у детей выражается беспокойством, зажмуриванием и плачем;
- птоз (опущение века) – ложный птоз обусловлен невозможностью прямого взгляда (на лицо собеседника) и взгляда вверх из-за светобоязни;
- зрачковые реакции– прямую и содружественную реакцию зрачков на свет при ВА проверить невозможно из-за отсутствия радужной оболочки глаза. Визуально создается впечатление отсутствия зрачка или очень темных глаз;
- нарушение мышечного тонуса– при аниридии наблюдается мышечная гипотония, проявляющаяся в снижении сопротивления в суставах при пассивных движениях; увеличении амплитуды движений в суставах; задержке моторного развития;
- нарушение сна– позднее отхождение ко сну, трудности при засыпании, тревожный и кратковременный сон. Часто ритуал засыпания сопровождается привычными патологическими действиями (яктация – раскачивание, дети теребят одеяло, наматывают и перебирают волосы, грызут ногти).
При ВА также отмечается повышенная чувствительность к звуковым раздражителям, запахам. На первом году жизни у ребенка с этой нозологией нередко наблюдаются отклонения в психоэмоциональном развитии, отмечается запаздывание формирование комплекса оживления. Дети позже начинают фиксировать взгляд на предметах. Только к 3—5 месяцам формируется слежение за игрушкой, кратковременное, зависящее от освещения в помещении и цветовой характеристики предмета. Низкая острота зрения затрудняет узнавание лиц; ребенку с аниридией требуется дополнительное время для адаптации в новой обстановке. Однако при этом его познавательная активность сохранна. К особенностям поведения при ВА можно отнести осторожность, повышенную тревожность, пищевую привязанность к хорошо знакомым блюдам.
1. Клочко Н.А. Особенности неврологических нарушений при аниридии / методические рекомендации.
file:///C:/Users/user/Desktop/аниридия%20метод%20рек%20%20%20невропатолога%20Клочко%20Натальи.pdf.
Лечение: своевременное и комплексное
КОНСЕРВАТИВНОЕ ЛЕЧЕНИЕ
ПРИ ВРОЖДЕННОЙ АНИРИДИИ (ВА):1-6
- профилактика и лечение аниридийной кератопатии (АК) – с раннего возраста назначение медикаментозной терапии офтальмологическими препаратами (кератопротекторы без консервантов: гипромеллоза или гипромеллоза + декстран; натрия гиалуронат – при недостаточной эффективности), при АК и/или для профилактики роговичных осложнений – глазной гель декспантенол;
-
противоглаукомные препараты и миотические средства (гипотензивные средства; аналоги простагландинов, бета-адреноблокаторы, ингибиторы карбоангидразы, антихолинэргические средства, парасимпатомиметики и др.) при наличии глаукомы. Алгоритм назначения противоглаукомных препаратов и миотических средств (гипотензивных средств) при аниридийной глаукоме:
- препараты первой линии выбора при врожденной глаукоме – ингибиторы карбоангидразы и бета-адреноблокаторы;
- при высоком ВГД – комбинированные препараты; при недостаточной эффективности добавляется препарат из другой фармакологической группы (простагландины, парасимпатомиметики);
- нейротрофическое лечение – для сохранения и стимуляции зрительных функций проводится регулярно, 1—2 раза в год. Терапия включает ноотропные препараты, ангиопротекторы, поливитамины и проводится под контролем невролога.
ХИРУРГИЧЕСКОЕ ЛЕЧЕНИЕ
ПРИ ВРОЖДЕННОЙ АНИРИДИИ (ВА)1,6-9:
- Хирургическое лечение глаукомы при ВА:
- хирургическое лечение – срочное или наиболее раннее лечение при первых признаках декомпенсации глаукомы у детей с ВА для снижения ВГД;
- рекомендуемые хирургические методы формирования оттока водянистой влаги:
- операции, направленные на устранение органических препятствий на пути водянистой влаги к трабекуле (гониотомия, в т.ч. с гониопунктурой; трабекулотомия ab interno)
- вмешательства фистулизирующего типа, предусматривающие формирование нового канала из передней камеры глаза наружу, в интрасклеральное пространство – диатермогониопунктура, микродиатермогониопунктура, трабекулотомия ab externo, операция гониодиализа с трабекулотомией ab externo, синустрабекулэктомия (СТЭ) и ее модификации).
- модифицированная СТЭ с аппликацией рекомендована для уменьшения рубцевания тканей и/или применение имплантов (дренажные устройства) детям с ВА и глаукомой при отсутствии эффекта от иных операций для нормализации офтальмотонуса или в качестве операции первого выбора при наличии факторов риска;
- при неэффективности хирургического лечения, направленного на улучшение оттока ВГЖ, рекомендованы методы, направленные на уменьшение секреции водянистой влаги: лазерная транссклеральная циклокоагуляция, диатермокоагуляция.
- Хирургическое лечение катаракты при ВА:
- факоаспирации или аспирации-ирригации врожденной катаракты с/без имплантации интраокулярной линзы (ИОЛ) в зависимости от характера помутнения хрусталика и наличия сопутствующей патологии глаза;
- современная технология хирургии катаракты включает: тоннельные роговичные или корнеосклеральные микро-разрезы с последующей шовной фиксацией основного разреза, выполнение парацентезов на 3 и 9 часах; использование современных вискоэластиков для защиты эндотелия роговицы; применение дифференцированных методик переднего к апсулорексису с формированием минимального диаметра (менее 5 мм) для более стабильной фиксации ИОЛ в капсульном мешке; аппаратная или мануальная аспирация-ирригация хрусталиковых масс; эндокапсулярная имплантация гибких акриловых ИОЛ;
- рекомендовано проведение консервативного медикаментозного и при необходимости хирургического лечения у детей с ВА при развитии интра- и послеоперационных осложнений;
- всем детям с ВА в послеоперационном периоде – назначение противовоспалительных и антибактериальных препаратов для профилактики инфекции и воспаления.
P.S. Осложнения хирургического вмешательства при ВА – аниридийный фиброзный синдром (АФС) (фиброзный рубцовый процесс, ассоциированный с развитием гипотонии, снижающий зрительные функции и в ряде случаев приводящий к субатрофии глаза); синдром мелкой камеры, цилиохориоидальная отслойка (ЦХО), гифема, макулярный отек.
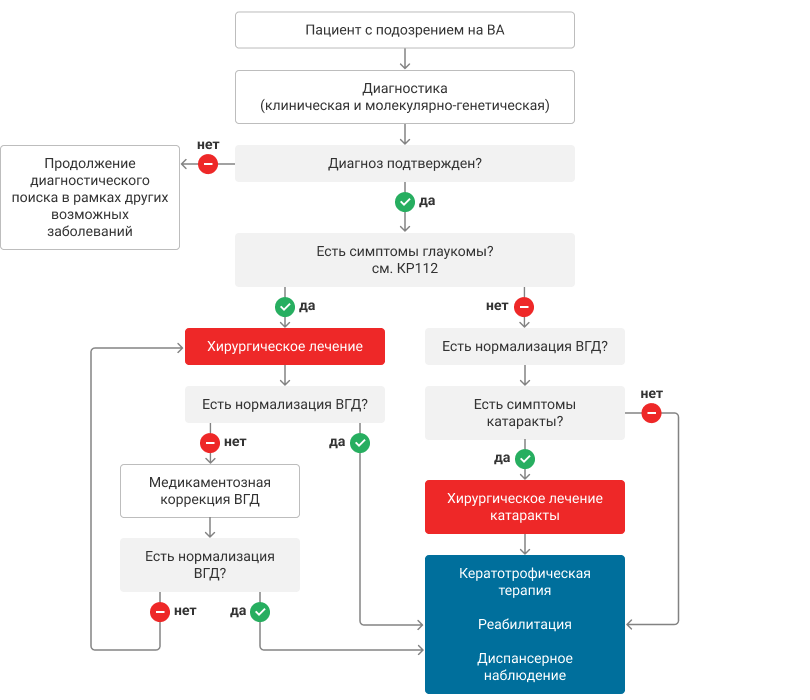
Рисунок. Алгоритм действий врача при врожденной аниридии1
1. Аниридия врожденная. Клинические рекомендации. https://cr.minzdrav.gov.ru/preview-cr/740_1?ysclid=m5yztw8h1o73408754 (Дата обращения: 15.01.2025).
2. Seitz B., Kasmann-Kellner B., Viestenz A. Stage-related therapy of congenital aniridia. Ophthalmologe. 2014, 111 (12), p. 1164—1171.
3. Lopez-Garcia J.S., Garcia-Lozano I., Rivas L., Martinez-Garchitorena J. Congenital aniridia keratopathy treatment. Arch. Soc. Esp Oftalmol. 2006, 81 (8), p. 435—444.
4. Coppens G., Stalmans I., Zeyen T., Casteels I. The safety and efficacy of glaucoma medication in the pediatric population. J. Pediatr. Ophthalmol Strabismus. 2009, 46 (1), p. 12—18.
5. Катаргина Л.А., Тарасенков А.О., Мазанова Е.В. Ретроспективный анализ применения гипотензивных медикаментозных средств при врожденной глаукоме на базе детского хирургического отделения // Российская педиатрическая офтальмология. № 1, 2013, с. 8—12.
6. Хватова А.В., Яковлев А.А., Теплинская Л.Е. Врожденная глаукома: современный взгляд на патогенез и лечение. Зрительные функции и их коррекция у детей. «Врожденная глаукома: современный взгляд на патогенез и лечение». Медицина. 2005, c. 319—344.
7. Okada K., Mishima H.K. et al. Results of filtering surgery in young patients with aniridia. Hiroshima J. Med. Sci. 2000, 49 (3), p. 135—138.
8. Swanner J.C., Samant M., Chauhan B.K., Lathrop K.L., Nischal K.K. Congenital aniridia: etiology, manifestations and management. Expert Review of Ophthalmology. 2016, 11 (2), p. 135—144, DOI: 10.1586/17469899.2016.1152182.Walton D.S., Chen T.C. Pre- vention of aniridic glaucoma with goniosur- gery. Int Ophthalmol. Clin. 2004, 44 (1), p. 67—71.
9. Reinhard T., Engelhardt S., Sundmacher R. Black diaphragm aniridia intraocular lens for congenital aniridia: long-term follow-up. J. Cataract Refract Surg. 2000, 26 (3), p. 375—381.
Медицинская реабилитация и наблюдение

Патология требует диспансерного наблюдения детским офтальмологом, регулярных и тщательных осмотров, раннего выявления признаков прогрессирования осложнений, а также своевременного взвешенного подхода к вмешательствам.1-2
При подтверждении диагноза изолированной аниридии детям в возрасте от 0 до 10 лет необходимы:1-6


