















Российские ученые классифицировали данные из крупнейшей базы онкогенных мутаций
На основании геномных данных 10 тыс. пациентов исследователи из лаборатории разработки инновационных лекарственных средств и агробиотехнологий Физтех-школы биологической и медицинской физики МФТИ подсчитали количество онкогенных мутаций молекулярных и функциональных типов в разных видах рака у пациентов различных демографических и клинических групп. Главной целью классификации было разделение мутаций на значимые и незначимые, чтобы в дальнейшем упростить подбор необходимой терапии, сообщили «МВ» в пресс-службе МВТИ. Результаты исследования опубликованы в журнале PLOS Genetics
«Смертность от рака составляет примерно половину случаев общей смертности, а мутации и различные хромосомные дефекты считаются главной причиной и одновременно механизмом развития рака. Мы анализируем их, чтобы выявлять мишени для потенциальных терапевтических воздействий, чтобы знать, на какие гены, на какие белки воздействовать», — пояснил один из авторов исследования, старший научный сотрудник лаборатории разработки инновационных лекарственных средств и агробиотехнологий МФТИ Алексей Беликов.
Для классификации мутаций, в том числе на значимые и незначимые, ученые разработали четыре биоинформатических алгоритма, с помощью которых обработали геномные данные из самой крупной базы данных пациентов с онкологией — TCGA PanCanAtlas.
Как уточнил Беликов, один из четырех алгоритмов выявляет онкогенные хромосомные дефекты, возникающие в опухолях. Причем делает это лучше, чем существующая до сих пор единственная в мире подобная американская программа, и выявленные дефекты подтверждаются другими научными исследованиями.
Среди большого количества мутаций и различных хромосомных аномалий новые алгоритмы позволили определять значимые для развития раковой опухоли. В частности, было выявлено, что в некоторых видах рака на развитие опухоли влияет только одна онкогенная мутация, а в некоторых — два десятка.
«Если взять образец опухоли конкретного пациента и отсеквенировать его, анализ может показать сотни мутаций, а для лечения придется подобрать ингибиторы сотни белков, что не представляется возможным, — отметил Беликов. — Наш анализ показывает, что в среднем значимыми оказываются 12 мутаций на опухоль, и это позволяет воздействовать именно на нужные белки, а не действовать вслепую».
Исследование позволило с высокой точностью определить количество различных типов онкогенных мутаций. Были проведены классификации по полу и возрасту пациентов, типу и стадии рака, а также по другим критериям.
Распределение онкогенных мутаций по типам рака
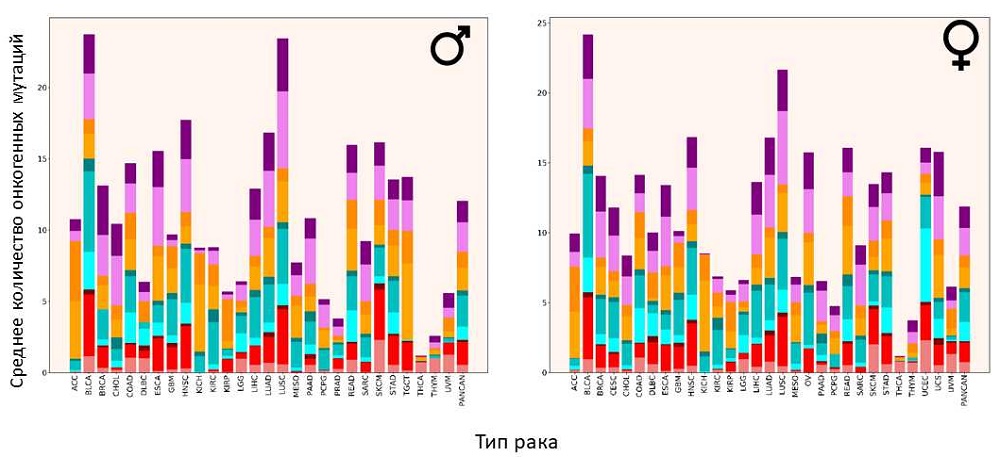 Источник: PLOS Genetics
Источник: PLOS Genetics
Распределение онкогенных мутаций по возрасту
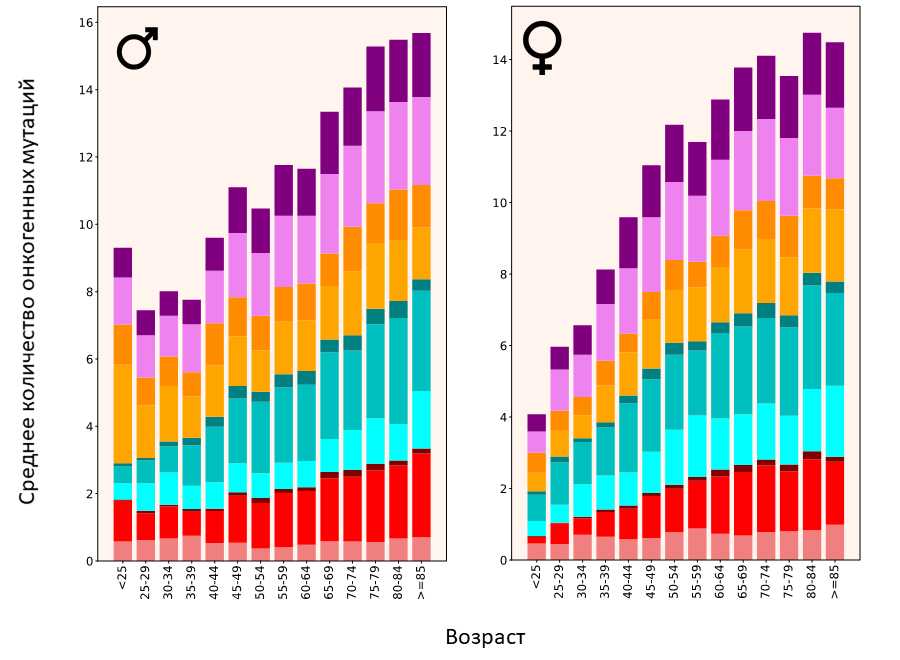
Источник: PLOS Genetics
Помимо ответов, проведенная классификация ставит большое количество вопросов и открывает перспективы для дальнейшей работы. Например, почему существует такая большая разница в количестве и составе онкогенных мутаций между типами рака? Почему для инициации рака щитовидной железы достаточно только одной онкогенной мутации, а при карциномах мочевого пузыря – несколько десятков? Почему у одних видов рака нет изменений в онкогенах, а у других нет изменений в опухолевых супрессорах? Объясняются ли эти различия разным тканевым микроокружением, к которому эти опухоли должны приспосабливаться? Почему тогда для некоторых пациентов с одним и тем же типом рака достаточно одной онкогенной мутации для развития обнаруживаемой опухоли, тогда как у других опухоли не диагностируются, пока не будут накоплены десятки мутаций?


Нет комментариев
Комментариев: 0