














1.1. Определение заболевания или состояния (группы заболеваний или состояний)
1.1.1. Определение эпилепсии
Эпилепсия – заболевание головного мозга, определяемая любым из следующих условий: 1) по крайней мере, два неспровоцированных (или рефлекторных) приступа, с интервалом > 24 ч; 2) один неспровоцированный (или рефлекторный) приступ и вероятность повторения приступов, близкая к общему риску рецидива (≥ 60%) после двух спонтанных приступов в последующие 10 лет; 3) диагноз эпилептического синдрома,
(≥ 60% - следует трактовать как высокую вероятность рецидива) [3].
1.1.2. Определение эпилептического статуса
Эпилептический статус – состояние пролонгированного приступа или повторяющихся приступов, в интервалах между которыми состояние больного не возвращается к исходному. Это результат отказа механизмов, ответственных за прекращение, либо инициация механизмов, ведущих к аномально пролонгированным приступам после временной точки t1 (время начала лечения), которые могут иметь долгосрочные последствия после рубежа t2 (время начала долгосрочных изменений), включающие нейрональную смерть, нейрональное повреждение, перестройку нейронных связей. Временные параметры: тонико-клонический эпилептический статус t1 – 5 мин, t2 – 30 мин, фокальный эпилептический статус t1 – 10 мин, t2 – более 60 мин, статус абсансов t1 – 10 - 15 мин, t2 – неизвестно [4].
1.2.Этиология и патогенез заболевания или состояния (группы заболеваний или состояний)
1.2.1. Этиология эпилепсии
Эпилепсия – полиэтиологичное заболевание. В соответствии с классификацией эпилепсий Международной противоэпилептической лиги 2017 года [5], все формы эпилепсии подразделяются по этиологии на 6 категорий:
· генетические;
· структурные;
· метаболические;
· инфекционные;
· иммунные;
· с неизвестной причиной.
Определение этиологии эпилепсии играет решающую роль в выборе тактики ведения и лечения пациента. В ряде случаев у пациента может быть сочетание нескольких этиологических факторов, например, структурного и генетического. В.А. Карлов рекомендует рассматривать этиологические факторы как факторы риска эпилепсии, которые могут быть реализованы только при наличии наследственного предрасположения [6].
Генетические эпилепсии
В генетическую группу включено большое количество заболеваний, хромосомных и генных, как моногенных, так и полигенных, при которых эпилепсия может быть единственным проявлением заболевания или она входит в структуру заболевания наряду с другими симптомокомплексами.
Наследственные эпилепсии - группа генетически гетерогенных заболеваний, возникающих в результате мутаций в генах, количественных или структурных перестройках хромосом. В зависимости от этиологии можно выделить три основные группы наследственных эпилепсий: моногенные заболевания и синдромы, хромосомные синдромы и мультифакторные эпилепсии. Существует несколько групп моногенных заболеваний, в структуре симптомокомплекса которых отмечаются судороги: изолированные моногенные эпилепсии, моногенные синдромы и пороки развития мозга, дегенеративные заболевания нервной системы, наследственные болезни обмена веществ. В настоящее время идентифицировано более 700 генов, мутации в которых приводят к возникновению моногенных судорог [21].
Изолированные моногенные эпилепсии включают группы: ранних эпилептических младенческих энцефалопатий, миоклонус-эпилепсий детского и юношеского возраста, генерализованных эпилепсий с фебрильными судорогами плюс, доброкачественных фебрильных судорог височных и лобных эпилепсий. Каждая из этих групп насчитывает несколько генетических вариантов, обусловленных мутациями в отдельных генах [22].
Возникновение эпилепсий наблюдается также у больных с количественными и структурными перестройками хромосом. Идентифицировано несколько сотен хромосомных синдромов, сопровождающихся судорогами.
Значительная доля заболеваний приходится также на мультифакторные эпилепсии, возникающие при совместном действии наследственных факторов и факторов внешней среды. Наследственные факторы, в виде полиморфизмов в нескольких генах, формируют предрасположенность к возникновению судорог, которая реализуется под действием факторов внешней среды (травмы, инфекции, стресс и др.).
Установление этиологического фактора наследственного заболевания или синдрома, в большинстве случаев, является сложной задачей, так как требует использования различных биохимических и молекулярно-генетических методов исследования. Однако, обнаружение гена или хромосомной перестройки, ответственных за их возникновение, необходимо не только для уточнения диагноза, определения характера течения заболевания и эффективности его терапевтической и хирургической коррекции, но и для расчета риска рождения больного ребенка в отягощенной семье и планирования профилактических мероприятий [23].
Особенности клинических проявлений идиопатических и синдромальных вариантов моногенных эпилепсий.
Предположить наличие моногенного варианта идиопатических эпилепсий возможно в следующих случаях:
- наличие нескольких членов семьи, страдающих эпилепсией;
- отсутствие провоцирующего фактора возникновения судорог (инфекции, травмы и др);
- фармакорезистентность судорог;
- отсутствие значимой очаговой неврологической симптоматики у пациентов с судорогами.
В большинстве случаев у пациентов с моногенными вариантами эпилепсий судороги возникают после периода нормального психомоторного развития, однако, в ряде случаев они возникают с рождения или даже во внутриутробном периоде.
У пациентов с моногенными синдромами, сопровождающимися судорогами, как правило, определяется специфический симптомокомплекс, при котором судороги являются одним из его симптомов.
Структурные эпилепсии
Подтверждённой структурной причиной эпилепсии считают изменения головного мозга, которые могут быть выявлены с помощью методов нейровизуализации и которые в совокупности с клиническими и нейрофизиологическими данными позволяют с высокой долей вероятности предположить их связь с возникновением эпилептических приступов [5, 6]. Связанные с эпилепсией структурные изменения могут быть приобретёнными (например, вследствие черепно-мозговой травмы или внутриутробной инфекции) или генетически обусловленными (например, нарушения развития коры). У отдельных пациентов возможно сочетание различных потенциально эпилептогенных структурных изменений головного мозга (например, склероза гиппокампа и фокальной кортикальной дисплазии).
Церебральные дизонтогенезии, или мальформации развития коры, по современным данным, являются наиболее частой причиной эпилепсии, особенно проявляющейся у детей и подростков, и представляют собой большой спектр нарушений, поражающий целиком обе гемисферы (например, лиссэнцефалия), либо распространенные билатеральные поражения (например, билатеральная узловая гетеротопия), либо целиком одну гемисферу (например, гемимегалэнцефалия), либо изолированные участки коры одного полушария (фокальные кортикальные дисгенезии). Тяжелые дизонтогенетические поражения мозга, проявляющиеся эпилепсией и слабоумием, были идентифицированы в начале прошлого столетия на основании патологоанатомических исследований [Alzheimer A., 1907]. Клинико-морфологические сопоставления позволили описать клиническую картину многих таких синдромов и диагностировать их в младенчестве и раннем детстве. Это относится прежде всего к болезни Штурге - Вебера, лиссэнцефалии, туберозному склерозу, гемимегалэнцефалии. В диагностике ряда других дисплазий, лежащих в основе эпилепсии, особенно фокальной корковой дисплазии, как и ряда гетеротопий, выдающуюся роль сыграла магнитно-резонансная томография (МРТ) [6]. Классификация нарушений кортикального развития представлена в таблице 1 [65].
Таблица 1. Классификация нарушений кортикального развития [525]
Группа I. Нарушения вследствие аномальной пролиферации нейронов и глии или апоптоза | |
|---|---|
I. A |
Микроцефалия |
I.B |
Мегалэнцефалии, включая гемимегалэнцефалию |
I.C |
Кортикальные дисгенезии с аномальной клеточной пролиферацией (ФКД II типа по классификации МПЭЛ, 2011). |
Группа II. Нарушения вследствие аномальной нейрональной миграции | |
II. A |
Гетеротопия серого вещества |
II. B |
Лиссэнцефалия |
II. C |
Подкорковая ленточная гетеротопия (агирия – пахигирия – ленточный спектр) |
II. D |
«Булыжниковая» мальформация |
Группа III. Нарушения вследствие аномалии постмиграционного развития (аномалии корковой организации) | |
III. A |
Полимикрогирия |
III. B |
Шизэнцефалия |
III. C |
Фокальные кортикальные дисплазии (ФКД I и III типов по класс. МПЭЛ, 2011). |
Фокальная кортикальная дисплазия (ФКД) представляет собой участок мальформации коры головного мозга, который может иметь различные размеры и локализацию. Локальные изменения коры могут проявляться изменениями самих клеток коры (например, цитомегалия нейронов, баллонные клетки), аномальным их расположением (гетеротопии нейронов в слоях неокортекса или в подкорковом белом веществе), а также дезорганизацией коры и полимикрогирией. Эксперты МПЭЛ выделяют три типа ФКД [10, 11].
Тип I характеризуется в первую очередь аномальной ламинацией неокортекса в виде: нарушения радиальной миграции клеток с образованием «микроколонн» нейронов (тип Ia), нарушения шестислойного строения коры и нечёткости границы серого и белого вещества (тип Ib) или их сочетанием (тип Ic). Для ФКД I типа не характерно наличие морфологически изменённых клеток, но могут присутствовать незрелые клетки малого диаметра или гипертрофические пирамидные клетки с нормальной морфологией вне 5 слоя коры. Поскольку плотность серого вещества значимо не изменяется, ФКД I типа в большинстве случаев не удаётся выявить методами нейровизуализации.
Тип II характеризуется, помимо грубого нарушения послойного строения коры, наличием дисморфических нейронов большого диаметра. ФКД II типа дополнительно разделяют на подтипы в зависимости от отсутствия (тип IIa) или наличия (тип IIb) баллонных клеток. ФКД II типа, особенно IIb типа, чаще можно выявить с помощью магнитно-резонансной томографии по таким признакам как локальное утолщение коры, нечёткость границы серого и белого вещества, изменение сигнала от серого и подкоркового белого вещества, а также нарушения строения борозд и извилин.
Тип III определяется как сочетание нарушения ламинации коры и иных значимых структурных изменений той же или соседней области головного мозга, а именно: склероза гиппокампа (тип IIIa), опухоли (тип IIIb), сосудистой мальформации (тип IIIc), других структурных изменений (тип IIId).
ФКД часто ассоциирована с наличием у пациента эпилептических приступов и сопровождается, особенно ФКД II типа, интериктальной эпилептиформной активностью на ЭЭГ, совпадающей по локализации с областью структурных изменений коры [11]. ФКД, как правило, не приводит к клинически значимому неврологическому дефициту, проявляясь исключительно эпилептическими приступами, семиология которых зависит от локализации поражения. Приступы могут дебютировать в любом возрасте и зачастую резистентны к медикаментозной терапии [10].
Склероз гиппокампа является самым частым структурным изменением головного мозга у пациентов с фармакорезистентной эпилепсией [7]. На МРТ склероз гиппокампа характеризуется уменьшением объёма гиппокампа, усилением сигнала на T2-взвешенных изображениях, а также нарушением своей внутренней архитектуры [8]. Патоморфологически эксперты МПЭЛ выделяют три типа склероза гиппокампа в зависимости от вовлечения различных анатомических сегментов гиппокампа: наиболее часто встречаемый тип 1 предполагает наличие склероза в той или иной степени во всех сегментах (от СА1 до СА4), тип 2 – преимущественное вовлечение в патологический процесс сегмента СА1 и тип 3 – преимущественное поражение СА4 сегмента [9]. В отдельный тип выделяют изменения гиппокампа по типу глиоза без патогистологических признаков склероза (т.е. без уменьшения числа нейронов), однако его клиническая значимость остаётся объектом изучения [9]. Склеротические изменения нервной ткани могут распространяться на соседние структуры за пределами гиппокампа, например, на миндалевидное тело и парагиппокампальную извилину [8].
Склероз гиппокампа зачастую сопровождается интериктальной эпилептиформной активностью на ЭЭГ, исходящей из данной области. Оперативное вмешательство на височной доле у пациентов со склерозом гиппокампа в 2/3 случаев приводит к освобождению от приступов с нарушением сознания как минимум в течение года [8].
Тем не менее, открытым остаётся вопрос, является ли склероз гиппокампа непосредственной причиной или следствием иного этиологического фактора эпилепсии. Так, склероз гиппокампа может сопровождаться другими структурными изменениями головного мозга, такими как фокальная кортикальная дисплазия, высокодифференцированные опухоли или сосудистые мальформации [8]. Известно также, что гиппокамп может претерпевать структурные изменения в результате продолжительной эпилептической активности во время эпилептического статуса, а также после черепно-мозговой травмы или воспаления [7].
Опухоли головного мозга могут являться причиной развития судорожных приступов при условии сдавления или вовлечения в патологический процесс коры головного мозга. Примерами могут служить менингиома или диффузная инфильтративно растущая глиома. В отдельную группу выделяют доброкачественные опухоли, ассоциированные с длительно присутствующей, фармакорезистентной эпилепсией – «long-term epilepsy associated tumors» (LEATs): к ним относятся в первую очередь ганглиоглиома и дисэмбриопластическая нейроэпителиальная опухоль, а также более редкие варианты, такие как, например, ангиоцентрическая глиома, изоморфная диффузная глиома, папиллярная глионейрональная опухоль [12,13]. Для группы LEATs характерны дебют эпилепсии в молодом возрасте (чаще до 13 лет) и височно-долевая локализация опухоли в большинстве случаев [12,13]. Появление опухолей группы LEATs связывают с нарушением развития мозга на ранних этапах, что объясняет их частую ассоциацию с кортикальными дисплазиями [13]. Остаётся неясным, что именно является причиной стойкой предрасположенности к возникновению эпилептических приступов и резистентности к противоэпилептическим препаратам – непосредственно опухоль или изменения прилежащей мозговой ткани [12].
Черепно-мозговая травма является наиболее частой причиной (фактором риска) приобретённой структурной эпилепсии. Приступы, возникшие в течение первых 24 часов после травмы, называют немедленными, а в течение 2 - 7 суток – ранними; они являются острыми симптоматическими приступами, спровоцированными травмой. Приступы, возникшие после 7 суток, называют поздними, их считают неспровоцированными эпилептическими приступами, т.е. проявлением эпилепсии [14,15].
Вероятность развития эпилепсии после черепно-мозговой травмы, по данным разных исследований, варьирует от 5 до 42% в зависимости от выборки и времени наблюдения [14]. Посттравматическая эпилепсия более чем в 90% случаев развивается в течение первых двух лет [15]. По прошествии 5 лет риск развития заболевания значительно снижается (< 1%), но вероятность развития неспровоцированных приступов сохраняется в течение 10 и даже 30 лет после черепно-мозговой травмы [14].
Факторами риска развития посттравматической эпилепсии являются тяжёлая черепно-мозговая травма, множественные ушибы головного мозга, повреждение твёрдой мозговой оболочки, вдавленный перелом черепа, внутричерепное кровоизлияние, а также длительность потери сознания или амнезия более суток. Наличие ранних посттравматических приступов также может увеличивать риск развития эпилепсии [15].
Медикаментозная противоэпилептическая терапия может быть эффективна, но не во всех случаях удаётся достичь ремиссии [14]. Часто, примерно в 1/3 случаев посттравматической эпилепсии, на магнитно-резонансной томографии кроме посттравматических изменений выявляется также склероз гиппокампа [15].
Перинатальные поражения ЦНС (антенатальные, натальные и ранние постнатальные) являются частыми этиологическими факторами развития эпилепсии у детей. Гипоксически-ишемические поражения ЦНС вследствие перинатальных факторов включают: внутриутробные инфекции, перинатальные инсульты, паренхиматозные кровоизлияния, билирубиновую энцефалопатию, поствакцинальные поражения ЦНС, наследственные болезни метаболизма [65].
Перинатальные поражения – наиболее частая причина неонатальных судорог, которые связаны главным образом с асфиксией плода (развитием гипоксически-ишемической энцефалопатии) и механической травмой головного мозга, нередко сопровождающихся внутричерепными кровоизлияниями [6].
Инсульт и его последствия являются одной из основных причин (факторов риска) эпилепсии среди лиц старшего возраста. В зависимости от времени, прошедшего с момента инсульта, приступы разделяют на ранние, возникшие в течение первых 7 суток, и поздние, возникшие после 7 суток. Ранними считаются острые симптоматические приступы, спровоцированные локальными метаболическими изменениями и потому не являющиеся непосредственным проявлением эпилепсии. При этом, наличие ранних приступов увеличивает риск развития эпилепсии у пациента в дальнейшем. Поздние приступы, наоборот, считают проявлением приобретённой предрасположенности головного мозга к возникновению эпилептических приступов, т.е. проявлением постинсультной эпилепсии [15,16].
Распространённость постинсультной эпилепсии достигает 12 - 15%, по данным разных исследований, но различается в зависимости от методологии исследования и длительности наблюдения [16,17]. Помимо наличия ранних приступов, факторами риска развития постинсультной эпилепсии являются: возраст до 65 лет, гипонатриемия, злоупотребление алкоголем в анамнезе, геморрагический тип инсульта, вовлечение коркового вещества, височнодолевая локализация поражения, а также тяжёлый неврологический дефицит в дебюте инсульта [16,18]. Фокальная эпилептиформная активность на ЭЭГ также является прогностически неблагоприятным фактором развития постинсультной эпилепсии [15].
Эпилептические приступы, ассоциированные с инсультом, в большинстве случаев поддаются медикаментозному контролю, однако до 25% пациентов не достигают ремиссии [15]. Эффективность профилактического назначения противоэпилептических препаратов в настоящее время не доказана [16].
Метаболические эпилепсии
Неонатальные судороги могут возникать при различных врожденных нарушениях метаболизма: органических ацидуриях, аминоацидопатиях, дефектах ферментов дыхательной цепи, расстройствах метаболизма пирувата, нарушениях обмена ẞ - окисления жирных кислот, расстройствах метаболизма карнитина и др. Многие метаболические причины развития эпилепсии также обусловлены генетически. В таблице 2 представлены курабельные наследственные метаболические заболевания, которые нельзя пропустить [65].
Таблица 2. Наследственные метаболические заболевания [526, с добавлением]
1. |
Дефицит В6 и фолиевой кислоты |
|---|---|
2. |
Дефицит транспорта глюкозы, тип I (болезнь Де Виво) |
3. |
Синдром гиперинсулинизма с аммониемией |
4. |
DEND (задержка развития, эпилепсия, неонатальный диабет) |
5. |
Гиперэкплексия |
6. |
Нарушение синтеза креатинина |
7. |
Дефицит биосинтетазы серина |
8. |
Биотинидазная недостаточность |
9. |
Дефицит фолата мозга |
10. |
Нарушение синтеза биоптерина |
11. |
Дефицит орнитинтранскарбамилазы (нарушение цикла мочевой кислоты) |
Инфекционные эпилепсии
Под инфекционной этиологией эпилепсии понимают известную инфекцию, ключевым проявлением которой являются приступы. Эпилепсия в данном случае возникает вследствие нейроинфекции и характеризуется формированием стойкой предрасположенности мозга к возникновению приступов, а не только судорожными приступами в остром периоде инфекционного заболевания. Обусловленные нейроинфекцией изменения головного мозга также могут быть структурными [5].
Примерами нейроинфекций, способных привести к развитию эпилепсии, являются клещевой энцефалит, вирус Зика, цитомегаловирусная инфекция, вирус иммунодефицита человека, туберкулёз. К инфекционным эпилепсиям относят также эпилепсии, развивающиеся при инвазионных заболеваниях, например, при цистицеркозе, токсоплазмозе, эхинококкозе [5].
Иммунные эпилепсии. Причиной иммунной эпилепсии считают иммунное расстройство, основным проявлением которого являются приступы, и которое непосредственно приводит к развитию эпилепсии [5]. В большинстве случаев данным иммунным расстройством является аутоиммунный процесс, триггером которого служат онкологическое заболевание или инфекция, в том числе вирусный энцефалит [19]. Приступы, возникающие в результате аутоиммунного энцефалита, зачастую могут быть первым, преобладающим или даже единственным его проявлением и возникают с частотой от 33% до 100% случаев в зависимости от антигена [20]. Однако, далеко не всегда аутоиммунный энцефалит приводит к развитию эпилепсии как хронического заболевания, и часто приступы прекращаются после завершения острого периода болезни, который может длиться несколько месяцев. Таким образом, диагноз эпилепсии рекомендуют подтверждать после длительного наблюдения пациента с продолжающимися приступами, например, в течение 12 месяцев [20].
Различают аутоиммунные энцефалиты, характеризующиеся наличием антител: 1) к поверхностным клеточным антигенам, 2) к внутриклеточным антигенам (опосредован Т-клеточным иммунитетом). В первом случае риск формирования стойкой предрасположенности к возникновению эпилептических приступов после разрешения энцефалита, как правило, низкий, за исключением анти-LGI1 и анти-GABAaR энцефалитов. Во втором случае вероятность развития эпилепсии в исходе энцефалита, напротив, высокая. Примерами энцефалитов с антителами к внутриклеточным антигенам являются некоторые паранеопластические энцефалиты (анти-Hu, анти-Yo, анти-CRMP-5) и анти-GAD65 энцефалит [19,20].
Однако, несмотря на выдающиеся достижения в диагностике этиологии эпилепсии (прежде всего – высокоразрешающие методы нейровизуализации и генетического тестирования), большое количество форм проходят под рубрикой «неизвестная причина» (по старой терминологии «криптогенная эпилепсия»). Процент неустановленной этиологии колеблется в широком диапазоне от 20% до 64% всех случаев. По данным клинико-эпидемиологического исследования в РФ, фокальная эпилепсия неуточненной этиологии встречалась примерно в 34% случаев [45]. Процент выявляемости этиологических факторов эпилепсии у детей достоверно выше, чем у взрослых, несмотря на большее их разнообразие [527].
1.2.2. Патогенез эпилепсии
Патогенез эпилепсии не является единым для всех форм заболевания, хотя имеются общие универсальные звенья. Патогенез эпилепсии любого типа включает процесс эпилептогенеза: постепенное развитие судорожной активности и стадию сформировавшейся эпилепсии, причем эпилептогенез может продолжаться и при развившейся эпилепсии (рис. 1).
Рис. 1. Периодизация основных блоков патогенеза приобретенной эпилепсии
Общим признаком, характерным для патогенеза всех форм эпилепсии, является судорожная активность, вызванная пароксизмальными разрядами групп нейронов в результате избыточного возбуждения и/или недостаточного торможения. Возникшая в определенном участке мозга избыточная электрическая активность распространяется в соседние зоны, а также может передаваться к мышцам, вызывая конвульсии. Как правило, распространение возбуждения в подкорковые, таламические, стволовые и спинальные структуры соответствует тонической фазе судорожного приступа, а последующий тормозной импульс из таламуса прерывает тоническую фазу, которая сменяется спорадическими вспышками электрической активности в клонической фазе. В настоящее время единственным приемлемым биомаркером эпилептогенеза следует признать патологические высокочастотные осцилляции [24].
В формирование судорожной активности вносят вклад возбуждающие и тормозные постсинаптические потенциалы, изменения потенциалзависимых ионных каналов, а также изменения локальных концентраций ионов. Основным возбуждающим нейромедиатором является глутамат, реализующий свое действие через 2 типа глутаматных рецепторов: ионотропные, опосредующие быструю синаптическую трансмиссию (глутаматзависимые АМРА-, каинатные и NMDA-ионные каналы), и метаботропные, опосредующих медленную синаптическую трансмиссию (связаны с G-белками и регуляцией вторичных посредников цAMФ и фосфолипазы C). Основным тормозным нейромедиатором в ЦНС является гамма-аминомасляная кислота (ГАМК), имеющая в ЦНС 2 типа рецепторов: GABAA, постсинаптические специфичные, сопряженные с CI- -каналами и GABAB, пресинаптические ауторецепторы, снижающие высвобождение медиатора за счет снижения притока Са++ и сопряженные с постсинаптическими G-белками, что способствует повышению тока K+.
Клеточные механизмы генерации избыточного возбуждения при судорожной активности на уровне ионного равновесия включают токи Na+ и Ca++ в клетки за счет избытка нейромедиаторов глутамата и аспартата и изменения свойств их рецепторов. Недостаточное торможение за счет потока CI- в клетку и K+ из клетки обусловлено недостатком ГАМК или изменением свойств соответствующих рецепторов. К нейронным факторам, регулирующим возбудимость нейронов, относятся тип ионных каналов, их число и распределение на мембране нейрона, посттрансляционные модификации каналов (напр. фосфорилирование), активация систем вторичных посредников, влияющих на функционирование каналов (напр. G-белков), модуляция экспрессии генов ионных каналов. Некоторые формы эпилепсии тесно связаны с каналопатиями, являющимися результатом наследственных мутаций лиганд-зависимых ионных каналов (т. е. ионотропных рецепторов) и потенциалзависимых ионных каналов. Выявлены также типы приобретенной аутоиммунной эпилепсии, связанной с образованием антител против калиевых, натриевых и хлоридных каналов, а также изменением экспрессии каналов после судорог. При этом различные мутации одного и того же гена могут вызывать совершенно разные типы судорог и эпилепсии [23]. К синаптическим факторам, модифицирующим возбудимость нейронов, относятся изменения экспрессии лигандзависимых ионотропных каналов, посттрансляционные изменения в таких каналах, ремоделирование локализации или конфигурации синапсов, изменение синаптических функций щелевых контактов. Несинаптические (внешние) факторы модификации возбудимости нейронов включают изменения внеклеточных концентраций ионов, изменения внеклеточного пространства, модуляцию метаболизма нейромедиаторов или их захвата клетками глии. Механизмы генерации гипервозбудимости на сетевом уровне включают аксональный спрутинг возбуждающих нейронов, потерю тормозных нейронов, потерю возбуждающих нейронов, осуществляющих контроль тормозных нейронов, изменения импульсной активности нейронов (например при каналопатиях).
К общим признакам патогенеза эпилепсии относится воспаление, которое может быть связано с инфекцией или обусловлено нарушениями иммунной системы. Связь нейровоспаления и патогенеза эпилепсии, в т. ч. его важная роль в период эпилептогенеза, прослежена при различных формах эпилепсии в клинике и эксперименте [25]. Считается, что хронизации воспалительного процесса при эпилепсии способствуют активация микроглии и астроглиоз, сопровождающиеся повреждением нейронов. Одним из триггеров воспаления в ЦНС является повреждение гематоэнцефалического барьера, гематоликворного и ликвороэнцефалического барьеров, а также относительно автономного иммунного барьера мозга. В нейровоспалении и противовоспалительной защите мозга принимает участие система провоспалительных и противовоспалительных цитокинов, которые могут обладать как про-, так и антиконвульсантной активностью. Дисбаланс цитокиновой системы с преобладанием провоспалительных компонентов (интерлейкина 6, интерлейкина 1β, фактора некроза α) описан как у пациентов с эпилепсией, так и в различных моделях на животных и связан в первую очередь с активацией микроглии, а затем астроцитов. Активация цитокиновой системы как в количественном, так и в качественном отношении зависит от типа и выраженности судорожной активности, а также периода эпилепсии. Провоспалительные цитокины могут быть вовлечены в развитие гиперсинхронности нейронов и гипервозбудимости головного мозга за счет разных механизмов, в том числе взаимодействия с возбуждающими и тормозными нейромедиаторными системами, системой оксида азота в глиальных клетках и нейронах и сигнальными каскадами, приводящими к нейродегенерации и гибели нейронов. Некоторые из параметров магнитно-резонансной томографии коррелируют с наличием патологических высокочастотных осцилляций, могут косвенно отражать текущий воспалительный процесс в головном мозге и стать возможными биомаркерами эпилептогенеза. Общими патогенетическими механизмами развития эпилепсии, наряду с нейровоспалением и сопутствующим глиозом, являются также дисбаланс активных форм кислорода и окислительный стресс, нарушения систем антиоксидантной защиты (например, системы глутатиона). Дисфункция митохондрий сопровождается избыточной генерацией супероксидного анион-радикала и развитием окислительного повреждения ключевых молекул и клеточных органелл на фоне дефицита энергетических субстратов, в первую очередь аденозинтрифосфата (АТФ).
Медиальная височная эпилепсия (МВЭ), наиболее часто встречающаяся форма эпилепсии, подразумевает эпилептический синдром, при котором судорожная активность возникает из височной доли при активном вовлечении гиппокампа. Именно на МВЭ сфокусирована существенная часть фундаментальных исследований патогенеза эпилепсии, которые включают в первую очередь изучение патологии и патофизиологии гиппокампа при эпилептогенезе [26]. Возможные механизмы отложенного эпилептогенеза активно обсуждаются в рамках нескольких гипотез. Одна из них, модель киндлинга, предполагает, что повторяющиеся субконвульсивные стимулы, приводящие к последующим электрическим разрядам (afterdischarges), могут в конце концов приводить к развитию спонтанных судорог (эпилепсии). Другая модель рассматривает патофизиологию и изменения сетей гиппокампа при эпилептогенезе. Эта модель непосредственно связана с эпилепсией височной доли, но может быть полезна и для понимания развития других типов эпилепсии. Следует учитывать, что гиппокамп, а именно его зубчатая извилина, является еще и нейрогенной нишей, в которой образование новых нейронов (нейрогенез) продолжается в течение всей жизни. Зубчатая извилина выполняет функцию привратника (фильтра); в норме тормозная иннервация гранулярных клеток доминирует над возбуждающей, что позволяет зубчатой извилине контролировать возбуждение, при этом иннервация тормозных ГАМК-ергических интернейронов гранулярными клетками по механизму отрицательной обратной связи контролирует возбудимость гиппокампа.
Установлено, что при эпилептогенезе существенно усилено образование новых нейронов в зубчатой извилине гиппокампа. Эпилептиформная активность возникает, когда зубчатая извилина не может выполнить свою функцию фильтра возбуждения. Причиной этого может быть формирование аберрантных нервных сетей за счет вызванной нарушенным нейрогенезом реорганизации связей между гранулярными клетками. Предполагается, что именно формирование рекуррентных возбуждающих связей в процессе эпилептогенеза нарушает функцию зубчатой извилины. При этом аномальная интеграция новорожденных гранулярных клеток в гиппокампальные сети происходит за счет прорастания аксонов мшистых волокон, прорастания базальных дендритов в хилус, где они образуют синаптические контакты с мшистыми волокнами, миграции гранулярных клеток в хилус с нарушением морфологии гранулярного слоя. Эти изменения нарушают функционирование гиппокампа, способствуя существованию проэпилептогенных нервных сетей. Нейродегенерация и снижение нейрогенеза гиппокампа являются одним из общих патогенетических механизмов, лежащих в основе эпилептогенеза. Как и при ряде других неврологических и психических заболеваний, эти события тесно связаны с дисрегуляцией нейротрофинов, в частности, нейротрофического фактора мозга (с англ. Brain Derived Neurotrophic Factor, BDNF) [545]. Предполагается, что при лимбическом эпилептогенезе усиленная экспрессия BDNF вносит ключевой вклад в аберрантный нейрогенез и спрутинг мшистых волокон гиппокампа, способствуя тем самым длительной потенциации возбуждающей синаптической трансмиссии [27, 28]. Важно отметить, что на долю МВЭ приходится существенная часть пациентов с фармакорезистентной эпилепсией, при этом фармакорезистентность является проявлением как общей тяжести заболевания, так и результатом глубокого дисбаланса между многокомпонентной эпилептической и противоэпилептической системами головного мозга. Возможно, в основе фармакорезистентности лежат врождeнные или приобретeнные изменения активности белков-транспортеров гематоэнцефалического барьера и/или чувствительность молекулярных мишеней противоэпилептических препаратов.
У пациентов с опухолями мозга за счет снижения перфузии в области опухоли и усиления метаболизма возникает гипоксия, вызывающая ацидоз и нарушения окислительного энергетического метаболизма, что приводит к набуханию клеток глии и повреждению окружающей ткани. Возникающий дисбаланс между возбуждением и торможением приводит к судорожной активности за счет повышенного внеклеточного уровня глутамата до нейротоксических значений. При глиоме эпилептическая активность возникает вне опухоли в районе околоопухолевой границы, где повышен уровень глутамата. Активность рецепторов ГАМК понижена, что также вносит вклад в развитие избыточного возбуждения [29].
Механизмы эпилептогенеза в результате ЧМТ сводятся к вопросу о причинах развития склонности к повторяющимся неспровоцированным судорожным приступам при отсутствии явных патологических провоцирующих факторов в позднем периоде ЧМТ. Первичные повреждения при ЧМТ включают в себя острую клеточную гибель, нарушение гематоэнцефалического барьера; они приводят к деполяризации нейронов, выбросу возбуждающих нейромедиаторов и повышению экстраклеточной концентрации К+, а в конечном итоге к гиперсинхронизации нейронов, что проявляется острыми судорожными приступами у животных и, вероятно, у человека [30]. К клеточной гибели приводит острый чрезмерный выброс глутамата и аспартата, вызывающий активацию NMDA-рецепторов, вход Na+ и Ca++ в клетку, выбросу K+, апоптоз и некроз нейронов в результате эксайтотоксичности[31]. Вторичное повреждение в результате ЧМТ связано с активацией процессов отложенной клеточной гибели, нейровоспаления, глио- и ангиогенеза. Многие из этих процессов вовлечены в эпилептогенез: гибель нейронов, глиоз, нейровоспаление, нарушение гематоэнцефалического барьера, нарушение возбудимости нейронов, нарушенные ангиогенез и нейрогенез, изменение синаптической пластичности, перестройка нейрональных сетей, изменения экспрессии генов и эпигенетические модификации [32]. Помимо формирования прямого очага повреждения в коре, ЧМТ приводит к дистантной и вторичной гибели нейронов и активации глии в гиппокампе [33], в первую очередь ГАМК-ергических вставочных нейронов хилуса [34]. Нейровоспаление после ЧМТ присутствует как в остром периоде, обусловливая отёк и нейродегенерацию, так и в хроническом периоде [35]. Клеточный субстрат нейровоспаления в остром периоде в основном представлен микроглией, в хроническом большую роль играют астроциты. Синтезируемые иммунными клетками цитокины модифицируют функцию глутамат- и ГАМК-ергических рецепторов, ингибируют захват глутамата астроцитами, нарушают функцию потенциал-зависимых ионных каналов, ведут к повышению экстраклеточной концентрации К+, и всё это фо
1.3.1. Эпидемиология эпилепсии
Более 50 миллионов человек во всем мире страдают эпилепсией [41]. На эпилепсию приходится 13 миллионов лет жизни, скорректированных на инвалидность [42].
Согласно определению Международной противоэпилептической Лиги, наличие активной эпилепсии у пациента подразумевает прием противоэпилептических препаратов либо наличие приступов за последние 2 - 5 лет [43]. Стандартизированная по возрасту распространенность активной эпилепсии в мире по данным на 2016 год составляет 621,5 (540,1 – 737,0) на 100000. Распространенность активной эпилепсии увеличивается с возрастом, достигая максимума к 5 - 9 годам (374,8 [280,1 – 490,0]) и у людей старше 80 лет (545,1 [444,2 – 652,0])[42]. Заболеваемость эпилепсией в разных странах составляет в среднем 67.77 на 100000 человек в год (95% CI 56,69 – 81,03) [44].
По данным единственного российского масштабного клинико-эпидемиологического исследования 517624 человек 14 лет и старше в 14 регионах РФ (0,34% всего населения РФ) стандартизированное по возрасту значение распространенности (European Standard Million) составило 3,40 случая на 1000. Распространенность эпилепсии была выше: в Сибири и на Дальнем Востоке по сравнению с Европейской частью РФ, в сельской местности по сравнению с крупными городами. Возрастная структура заболеваемости отличалась от наблюдаемой в странах Европы и США – значения заболеваемости были ниже в старших возрастных группах[45].
Каждый год регистрируется 125000 смертей больных эпилепсией [46]. Стандартизированные показатели смертности пациентов с эпилепсией в странах с низким и средним уровнем дохода более чем в 2,5 раза, а в странах с высоким уровня дохода – в 2 - 7 раз - превышают общепопуляционные [47,48].
Преждевременная смертность больных эпилепсией обусловлена в том числе более частой травматизацией и суицидами, а также высоким уровнем соматической и психиатрической коморбидности [49–51].
Особое место среди причин смерти пациентов с эпилепсией занимает SUDEP (sudden unexpected death in epilepsy), или синдром внезапной смерти при эпилепсии, так как частота встречаемости этого синдрома среди молодых людей с эпилепсией, в особенности фармакорезистентной, по разным оценкам в 24 - 27 раз выше, чем в общей популяции [52].
1.3.2. Эпидемиология эпилептического статуса
Критический анализ популяционных исследований в различных регионах земного шара показал высокую вариабельность возникновения эпилептического статуса от 1,29 до 73,7 на 100000 взрослых [53]. По данным большинства исследований, риск развития эпилептического статуса выше у мужчин в сравнении с женщинами, а также у детей и людей старше 60 лет [54].
Согласно Международной классификации болезней 10-го пересмотра (МКБ-10), к эпилепсии относятся преимущественно коды G40 и G41 [55]:
•G40.0. Локализованная (фокальная, парциальная) идиопатическая эпилепсия и эпилептические синдромы с судорожными приступами с фокальным началом. Доброкачественная детская эпилепсия с пиками на ЭЭГ в центрально-височной области. Детская эпилепсия с пароксизмальной активностью на ЭЭГ в затылочной области;
•G40.1. Локализованная (фокальная, парциальная) структурная эпилепсия и эпилептические синдромы с простыми парциальными приступами.
Приступы без изменения сознания. Простые парциальные приступы, переходящие во вторично-генерализованные приступы;
•G40.2. Локализованная (фокальная, парциальная) структурная эпилепсия и эпилептические синдромы со сложными парциальными приступами.
Приступы с изменением сознания, часто с эпилептическими автоматизмами. Сложные парциальные приступы, переходящие во вторично-генерализованные приступы;
•G40.3. Генерализованная идиопатическая эпилепсия и эпилептические синдромы. Доброкачественная миоклоническая эпилепсия раннего детского возраста. Неонатальные судороги(семейные). Детские абсансы (пикнолепсия). Эпилепсия с большими судорожными приступами (grand mal) при пробуждении. Юношеская абсансная эпилепсия, миоклоническая эпилепсия (импульсивный малый приступ (petit mal)). Неспецифические эпилептические приступы: атонические, клонические, миоклонические, тонические, тонико-клонические;
•G40.4. Другие виды генерализованной эпилепсии и эпилептических синдромов. Эпилепсия с миоклоническими абсансами, миоклонико-атоническими приступами. Синдром Леннокса – Гасто. Структурная ранняя миоклоническая энцефалопатия. Синдром Веста;
· G40.5. Особые эпилептические синдромы.
Эпилепсия парциальная непрерывная (Кожевникова). Эпилептические приступы, связанные с употреблением алкоголя, применением лекарственных средств, гормональными изменениями, лишением сна, воздействием стрессовых факторов. При необходимости идентифицировать лекарственное средство используют дополнительный код внешних причин (класс ХХ);
•G40.6. Приступы grand mal неуточненные (с приступами petit mal или без них);
•G40.7. Приступы petit mal неуточненные без приступов grand mal;
•G40.8. Другие уточненные формы эпилепсии.
Эпилепсия и эпилептические синдромы, не определенные как фокальные или генерализованные;
•G40.9. Эпилепсия неуточненная;
•G41.0. Эпилептический статус grand mal;
•G41.1. Эпилептический статус petit mal;
•G41.2. Сложный парциальный эпилептический статус;
•G41.8. Другой уточненный эпилептический статус;
•G41.9. Эпилептический статус неуточненный;
•G83.8. Паралич Тодда;
•F80.3. Синдром Ландау – Клеффнера (приобретенная эпилептическая афазия);
•R56.0. Судороги при лихорадке;
•R56.8. Другие и неуточненные судороги;
•P90. Неонатальные судороги (исключено: семейные неонатальные судороги – G40.3).
Новая классификация эпилепсий МПЭЛ 2017 г. является многоуровневой и предназначена для применения в клинической практике (см. рис. 2) [5].
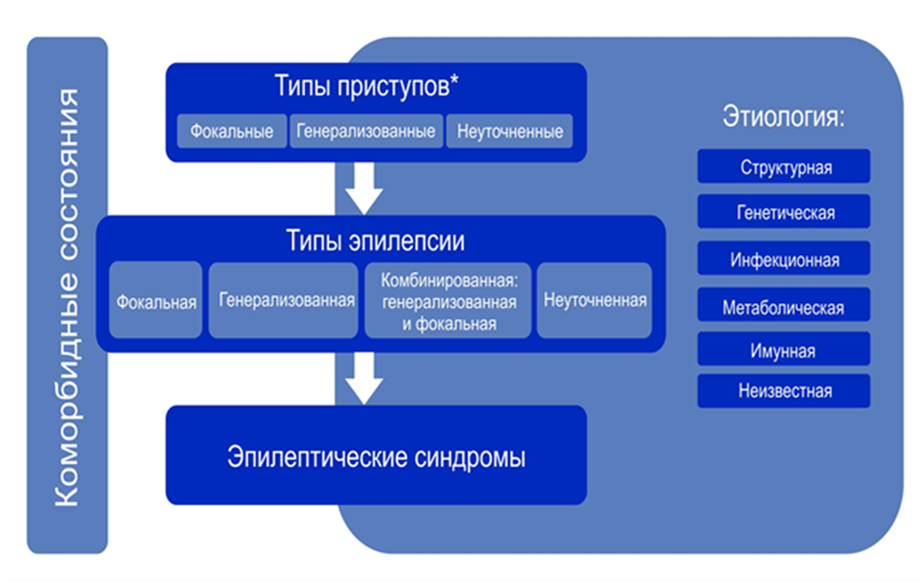
Рис.2. Схема классификации эпилепсий МПЭЛ 2017 г.
Данная классификация базируется на следующих принципах:
1. Определение типа приступов
2. Определение типа эпилепсии
3. Определение эпилептического синдрома
4. Определение этиологии эпилепсии
5. Определение коморбидных состояний
Классификация содержит несколько уровней, что обусловлено большой вариабельностью доступных методов обследования пациентов с эпилепсией в мире. На первом этапе (уровне) идет определение типа приступа: фокальный, генерализованный или с неизвестным началом (см. рис. 3). Фокальный эпилептический приступ определяется как приступ, исходящий из какой-либо области нейрональных сетей, ограниченных одним полушарием; эта зона может быть очень локальной или более распространенной. При этом возможно распространение на соседние зоны или переход на контралатеральное полушарие. Генерализованный эпилептический приступ определяется как приступ, исходящий из некоторой области головного мозга с быстрым распространением и билатеральным захватом нейрональных сетей. Неклассифицированный приступ определяется как приступ, который вследствие недостатка информации невозможно отнести к другим категориям в данный момент времени.

Рис. 3. Классификация эпилептических приступов МПЭЛ 2017 г.
В новой классификации МПЭЛ окончательно отказалась от термина вторично-генерализованные приступы, заменив его термином «билатеральные тонико-клонические приступы». Это связано с тем, что билатеральные тонико-клонические приступы не являются отдельным типом эпилептических приступов, а отражают распространение разряда из любых отделов коры и эволюцию любого типа фокальных приступов. Данная классификация вновь вернулась к необходимости оценки уровня сознания пациента во время фокальных приступов: в сознании, сознание нарушено, не известно.
На втором этапе (уровне) следует определиться с типом эпилепсии: фокальная, генерализованная или сочетанная фокальная и генерализованная, или неизвестная (unknown). Для генерализованной эпилепсии характерно наличие генерализованной спайк-волновой активности на ЭЭГ, спектр приступов, включая абсансы, миоклонические, атонические, тонические и тонико-клонические приступы. Диагноз ставится на основании клинических проявлений и типичных межприступных разрядов. Фокальные эпилепсии — это эпилепсии с одним или несколькими фокусами, а также эпилепсии с вовлечением одной гемисферы головного мозга. Для них характерен целый спектр клинических проявлений и фокальные эпилептиформные разряды на ЭЭГ. Сочетанные (комбинированные) генерализованные и фокальные эпилепсии — эпилепсии с фокальными и генерализованными типами приступов, при этом на ЭЭГ могут регистрироваться как фокальные, так и генерализованные разряды. Классическим примером такой эпилепсии является синдром Драве. Тип эпилепсии, диагностированный на втором этапе, может стать окончательным диагнозом, если клиницист не имеет возможности перейти к следующему уровню — выявлению эпилептического синдрома (как правило, в той ситуации, когда врач не имеет необходимых методов обследования пациента). В качестве примера приводится довольно распространенная ситуация височно-долевой эпилепсии без изменений на межприступной ЭЭГ. Достаточным в такой ситуации может считаться диагноз «фокальная эпилепсия неизвестной этиологии». Неклассифицированная эпилепсия (unknown) — эпилепсия, при которой невозможно определить, фокальная она или генерализованная, а данные ЭЭГ недоступны или мало информативны. Третий этап (уровень) заключается в установлении эпилептического синдрома. Эпилептический синдром представляет собой совокупность характеристик, включая тип приступа, данные ЭЭГ и нейровизуализации, он часто имеет возрастзависимый характер, провоцирующие факторы, хронозависимость и, в ряде случаев, определенный прогноз. Может отмечаться характерная коморбидность — интеллектуальные и психические нарушения. Синдром также может иметь ассоциированные этиологические, прогностические и терапевтические последствия. Он часто не соответствует этиологии эпилепсии, но определяет тактику лечения и наблюдения за пациентом. Существует достаточно много хорошо описанных эпилептических синдромов (детская абсансная эпилепсия, синдром Веста, синдром Драве и др.), классификация которых находится в разработке. Среди генерализованных эпилепсий выделяется общепризнанная и часто встречающаяся подгруппа — идиопатические генерализованные эпилепсии. К ним относятся детская абсансная, юношеская абсансная, юношеская миоклоническая эпилепсии и эпилепсия с изолированными генерализованными тонико-клоническими приступами. Греческий термин «idios» переводится как «сам», «свой», «личный» и подразумевает некую генетическую этиологию. Другая выделяемая группа синдромов — возрастзависимые (более точный перевод «самоограничивающиеся» от англ. self-limited) фокальные эпилепсии. К ним относятся доброкачественная эпилепсия детства с центротемпоральными спайками (синонимы — возрастзависимая эпилепсия с центротемпоральными спайками, роландическая эпилепсия), синдром Панайотопулоса, (синоним - эпилепсия детства с вегетативными симптомами), синдром Гасто (синоним -детская затылочная эпилепсия с визуальными симптомами), а также отдельные лобнодолевые, височнодолевые, и теменнодолевые эпилепсии с началом в подростковом и даже во взрослом возрасте. Четвертый этап (уровень) заключается в установлении этиологии эпилепсии. Согласно новой классификации, все эпилепсии делятся на структурные, генетические, инфекционные, метаболические, иммунные и с неизвестной этиологией. Этиологические факторы (факторы риска) подробно изложены в разделе 1.2.1. «Этиология эпилепсии». В дополнении к вышеизложенному следует отметить, что аутоиммунные механизмы, лежащие в основе таких эпилептических синдромов, как, например, синдром Расмуссена, формируют структурную эпилепсию. В свою очередь инфекционные агенты нередко индуцируют аутоиммунные процессы, которые приводят к повреждению мозга с клиническими проявлениями в виде эпилепсии (таким образом, эпилепсия сочетает инфекционную, аутоиммунную и структурную этиологию). Такое наблюдается в ряде приобретенных случаев синдрома Дайка — Давидоффа — Массона (Dyke — Davidoff — Masson syndrome), проявляющегося церебральной гемиатрофией, судорогами, гемипарезом, лицевой асимметрией и трудностями обучения, а также при эпилептическом синдроме, индуцированном фебрильной инфекцией (FIRES – сокращ. от англ. Febrile infection related epilepsy syndrome ) или так называемой разрушительной энцефалопатии детей школьного возраста (DESC – сокращ. от англ. Devastating epileptic encephalopathy in school-aged children).
Разрешение эпилепсии
Определение
Разрешение эпилепсии – это достижение определенного возраста у пациентов с зависящим от возраста эпилептическим синдромом (возрастзависимые эпилепсии детства) либо отсутствие эпилептических приступов в течение 10 лет у пациентов, не принимавших ПЭП не менее 5 последних лет.
Разрешение эпилепсии свидетельствует о том, что в данный момент времени эпилепсии у пациента нет, но нельзя с уверенностью исключить рецидив приступов в будущем. Риск рецидива зависит от формы эпилепсии, возраста, синдрома, этиологии, лечения и многих других факторов. Известна небольшая частота рецидивов после 5 лет без приступов, но нет данных о частоте рецидивов после 10-летнего бесприступного периода. 10-летний срок ремиссии был выбран в качестве критерия из-за предполагаемого очень низкого риска рецидивов. Более чем 10-летнее отсутствие приступов у пациентов, не получавших лечения последние 5 лет, по мнению экспертов рабочей группы МПЭЛ, свидетельствует об очень низком риске рецидива приступов [3, 481- 483].
1.6.1. Клиническая картина основных типов эпилептических приступов.
Дефиниция основных типов эпилептических приступов (ЭП) изложена в разделе Термины и определения [1,2, 484, 485].
1.6.2. Клиническая картина структурных фокальных эпилепсий (СФЭ)
Под структурными (симптоматическими) подразумевают формы эпилепсии с верифицированной причиной их развития. Большинство известных СФЭ развиваются на основе структурной аномалии головного мозга, опухолевого, травматического, гипоксически-ишемического, геморрагического или иного повреждения. Среди СФЭ в зависимости от локализации эпилептогенного очага выделяют лимбические (палеокортикальные) с очагом в области эмбриогенетически старых структур височной доли (гиппокампальный комплекс, поясная и зубчатая извилины и др.) и неокортикальные эпилепсии с очагом в области различных отделов новой коры [5]. Установление этиологии СФЭ - неотъемлемая часть адекватной тактики ведения пациента, особенно при решении вопроса о хирургическом лечении.
Особенности клинических проявлений фокальных структурных эпилепсий в раннем детском возрасте
Cемиология приступов у детей меняется с возрастом. По всей видимости процесс изменения семиологии отражает постепенность созревания нервной системы. Чем старше становится ребенок, тем больше клиническая картина эпилепсии напоминает таковую у взрослых пациентов.
В целом, фокальные приступы у детей первых лет жизни часто сопровождаются двусторонними клиническим проявлениями – могут выглядеть как двустороннее тоническое напряжение, как эпилептический спазм или двусторонний миоклонус. Без видео-ЭЭГ-мониторинга достаточно трудно определиться с точной привязкой эпилептического приступа к определенной доле головного мозга, то есть трудно определить эпилептогенную зону. Возможно, что исключение составляет дополнительная моторная зона коры; приступы, исходящие из нее, выглядят как асимметричное тоническое напряжение по типу позы «фехтовальщика».
Guadalupe Fernandez-Baca Vaca и соавт. в 2018 году на основании ретроспективного анализа видео-ЭЭГ-мониторинга у 1140 пациентов с эпилепсией определили следующие возрастные особенности фокальных приступов у детей в зависимости от возраста [57]. В возрасте от 0 до 3 лет по частоте фокальные приступы уступают генерализованным. Наиболее частым из фокальных является гипомоторный (по классификации 2017 года - не моторный с нарушением осознанности) фокальный приступ (он отмечается примерно у 20% детей первых трех лет жизни с эпилептическими приступами вообще); 50% этих приступов начинаются в затылочной доле, они латерализованы только в 25% всех случаев. У детей этой возрастной группы отсутствуют ауры и редко отмечаются автоматизмы (ороалиментарные автоматизмы с трудом отличаются от неэпилептических явлений в межприступном периоде).
В возрасте от 3 до 6 лет среди фокальных приступов начинают доминировать фокальные клонические и тонические типы (они отмечаются у 21% детей с эпилептическими приступами). Эпилептогенную зону по клиническим проявлениям и данным ЭЭГ удается определить в 60% всех случаев; если она фокальная, то моторный компонент будет контралатеральным. В этом возрасте появляются и становятся более отчетливыми ауры и автоматизмы, преимущественно ороалиментарные. Гипомоторные (немоторные с нарушением осознанности) приступы в этом возрасте не очень часты.
В возрасте от 6 до 10 лет сохраняются контралатеральные эпилептогенной зоне фокальные тонические и клонические приступы (22% от всех детей этого возраста с эпилептическими приступами). Отмечается невысокий процент пациентов с приступами нарушения осознанности без четких двигательных компонентов. Появляются пациенты с версивными и гипермоторными фокальными приступами.
В возрасте после 10 лет семиология приступов в целом соответствует таковой у взрослых пациентов с эпилепсией.
1.6.2.1. Структурная височная (височнодолевая) эпилепсия (СВЭ)
СВЭ – наиболее распространенная (25% всех форм СФЭ), полиэтиологичная, преимущественно структурная фокальная эпилепсия [58], развивающаяся на основе гиппокампального склероза (наиболее часто), фокальных кортикальных дисплазий (ФКД), дизэмбриопластических нейроэпителиальных опухолей (ДНЭТ), нередко ассоциированные со склерозом гиппокампа или ФКД [59]; комбинированные формы до 38% [6]. Более редкие причины - последствия перинатального повреждения, ЧМТ (19,5%) нейроинфекции (10%), лимбический энцефалит [60]. Выделяют две основные клинические формы СВЭ: лимбическую и неокортикальную.
Лимбическая (мезиотемпоральная) височная эпилепсия (МВЭ) - наиболее частая (60% всех СВЭ) и наиболее труднокурабельная форма, часто фармакорезистентная. Возраст дебюта – любой, чаще до 16 лет [7]. Более чем у половины больных дебюту МВЭ за несколько лет предшествуют атипичные фебрильные судороги [58] (характеристика в разделе «Фебрильные судороги»).
Клиника МВЭ [58]. Наиболее характерные типы эпилептических приступов (ЭП):
─ ЭП с сенсорным дебютом без расстройства сознания (стар. - изолированная аура). Часто с вегетативно–висцеральными абдоминальными проявлениями «с восходящим эпилептическим ощущением»; реже в форме обонятельных или вкусовых галлюцинаций, сновидных состояний (внезапно возникающие ощущения «снов наяву», грез, с чувством нереальности, ощущения «ранее виденного, слышанного, пережитого» или «никогда не виденного»); состояния деперсонализации (нарушение восприятия собственной личности); аффективных пароксизмов (немотивированное чувство страха, радости, приподнятости, легкости и экстаза);
─ фокальные ЭП с моторным дебютом и типичными автоматизмами (стар.: аутомоторные): изолированное выключение (или изменение) сознания без судорог с наличием ороалиментарных или жестовых (на стороне очага) автоматизмов и нередко дистонической установкой кисти на противоположной стороне;
─ фокальные ЭП с остановкой активности (стар.: диалептические);
─ билатеральные тонико-клонические приступы с локальным сенсорным или моторным началом.
Продолжительность ЭП - от 30 с до 3 мин; частота от единичных в месяц до нескольких раз в сутки. Характерны многообразие и сложность клинических проявлений МВЭ как в структуре самих приступов, так интериктальных непароксизмальных расстройств, среди которых доминируют нарушения познавательных функций (особенно у детей), аффективные расстройства, эпизодические психотические и дисфорические нарушения [6].
Неокортикальная (латеральная) височная эпилепсия встречается реже, дебютирует в любом возрасте. Для нее характерны фокальные ЭП в форме: слуховых (пароксизмальное возникновение ощущения шума, музыки, голосов) или зрительных (пароксизмальное появление сложных ярких панорамных зрительных образов, нередко с элементами воспоминания прошедших событий) галлюцинаций; приступов несистемного головокружения в сочетании с вегетативными проявлениями (бледность кожи, гипергидроз, тахикардия); пароксизмальной сенсорной афазии (при очаге в доминантном полушарии); «височных синкопе» (выключение сознания, обмякание и медленное падение без судорог) [61].
Важная клиническая особенность СВЭ – высокая частота двустороннего повреждения амигдало-гиппокампального комплекса [62], сочетание склероза гиппокампа, ДНЭТ, ФКД у одного больного, отсутствие интериктальных изменений или широкая топография интериктальных ЭЭГ нарушений (особенно у детей), выходящих за пределы височного региона [63], что создает определенные диагностические и тактические трудности.
1.6.2.2. Структурная лобная (лобнодолевая) эпилепсия (СЛЭ)
СЛЭ – полиэтиологичная СФЭ, чаще структурная, развивающаяся на основе ФКД, пороков развития головного мозга, последствий перинатальной энцефалопатии, опухоли, ЧМТ, ОНМК; реже инфекционная, иммунная, метаболическая или генетическая (синдром: гипермоторная эпилепсия с приступами, ассоциированными со сном). Дебют структурных форм СЛЭ возможен в любом возрасте.
Клиника. Выделяют 3 основные клинические и локализационные формы СЛЭ [64]: моторная, премоторная и префронтальная, определяющих особенности семиотики ЭП [65].
Моторная СЛЭ. ЭП развиваются из очага в области передней центральной извилины (моторная кора) проявляются клоническими судорогами лица, руки, ноги или гемиклоническими пароксизмами, Джексоновским маршем, иногда с последующим развитием паралича Тодда.
Премоторная СЛЭ. ЭП исходят из дополнительной сенсомоторной зоны, проявляются билатеральными асимметричными пароксизмами («по типу цифры 4»). Возможно начало с ощущения жжения с последующей вокализацией и тоническим напряжением руки контралатерально очагу.
Префронтальная СЛЭ. ЭП из дорсолатеральных и орбитофронтальных отделов лобной коры начинаются с тонического поворота глаз, а затем головы в противоположную очагу сторону; возможно развитие ЭП с вокализацией (вопли, стоны, сопение, кашель и др.) и сложными двигательными актами с гиперкинетическими проявлениями (педалирование, боксирование, сексуальные движения тазом); иногда встречается изолированная аура с нарушением когнитивных функций в виде «наплыва насильственных мыслей».
Все ЭП при СЛЭ могут протекать как при сохранном сознании, так и с выключением сознания, часто наблюдается трансформация в билатеральные тонико-клонические приступы (БТКП). Общая характеристика ЭП при СЛЭ: кратковременность (максимум минута), высокая частота жестовых автоматизмов, постуральных и моторных феноменов, серийных судорожных приступов и эпилептического статуса, наличие ингибиторных феноменов (застывание, замирание, внезапное падение), минимальная постиктальная спутанность, серийное циклолептическое течение и преимущественное возникновение в ночное время [6].
СЛЭ часто путают с генерализованными эпилепсиями вследствие персистирования приступов с генерализованным дебютом (псевдоабсансов, миоклонус век с абсансами), быстрой билатеральной синхронизацией возбуждения (на ЭЭГ) и моторной активности [66]. Иктальная ЭЭГ зависит от характера приступов. Наиболее часто констатируется низкоамплитудная быстрая активность в одном из лобных отведений, бифронтально или диффузно (Low Аmplitude Fast Activity – LAFA) [67].
Неврологический статус зависит от этиологии СЛЭ. Возможно формирование контралатерального гемипареза, атаксии, тугоподвижность и замедленность мыслительных процессов, признаки лобной психики, у детей - умственная отсталость, трудности обучения [65].
1.6.2.3. Структурная теменная (теменнодолевая) эпилепсия (СТЭ)
СТЭ - наиболее редкая форма в группе СФЭ, обычно структурная, в 1/3 всех случаев – опухолевого генеза [64], реже развивающаяся на основе ФКД, порэнцефалических кист, кортикальных дисплазий, ОНМК и др. Особо выделяют перинатальное ишемическое поражение на границе кровоснабжения ветвей средней и задней мозговых артерий («watershed lesion») с последующим развитием алигирии в зоне соединения теменной и затылочной долей [69].
Клиника. Дебют СТЭ возможен в любом возрасте. Семиотика ЭП связана с локализацией очага и распространением возбуждения на соседние регионы. Выделяют несколько клинических форм СТЭ (передняя, задняя, нижняя, парацентральная) [65]:
При передней СТЭ ЭП характеризуется в основном сенсорными феноменами (парестезиями, иногда болями или онемением, сенсорным Джексоновским маршем в контралатеральных конечностях) – «гемисенсорная» или «сенсомоторная» эпилепсия. Распространение возбуждения на рядом лежащие регионы сопровождается: присоединением зрительных иллюзий (на затылочную долю при задней СТЭ); головокружения, вегетативных абдоминальных симптомов, иногда с остановкой активности и автоматизмами (на височную долю при нижней СТЭ); моторных клонических феноменов (на моторную кору передней центральной извилины). У детей часто наблюдаются внезапно возникающие тонические билатеральные или аксиальные судороги, которые могут переходить в серии (распространение на лобную долю) [70].
При парацентральной СТЭ (очаг в области парацентральной дольки) ЭП проявляются сенсорными нарушениями в форме парестезии, жжения, болей в области гениталий, внутренних поверхностей бедер, ощущением оргазма, возможно появление БТКП, ЭП с нарушением речи при сохранности сознания, метаморфопсий, анозогнозии и аутотопагнозии в конечностях.
У пациентов с СТЭ нередко выявляется гемигипестезия по проводниковому типу, иногда в сочетании с легкой пирамидной недостаточностью контралатерально очагу. Возможны умеренные когнитивные расстройства. ЭЭГ выявляет эпилептиформную активность в теменных, теменно-затылочных или теменно-задневисочных отведениях, нередко – билатерально. Возможно появление феномена вторичной билатеральной синхронизации, а также продолженного регионального замедления. При позднем дебюте СТЭ необходимо, в первую очередь, исключить объемные образования. Прогноз зависит от этиологии СТЭ, частоты приступов и выраженности когнитивных нарушений.
1.6.2.4. Структурная затылочная (затылочнодолевая) эпилепсия (СЗЭ)
СЗЭ – редкая (5% всех эпилептических синдромов) обычно структурная, наиболее часто развивающаяся на основе: ФКД, последствий перинатальных энцефалопатий (затылочная улегирия), окципитальных кальцификатов, сосудистых аномалий (в т.ч. синдром Штурге – Вебера), MELAS-синдрома, опухолей или ОНМК в области затылочных долей. СЗЭ нередко относится к эпилепсиям со смешанной этиологией, генетической и структурной [69].
Заболевание дебютирует в любом возрасте. Иктальные клинические симптомы подразделяются на зрительные нарушения (простые и сложные зрительные галлюцинации, зрительные иллюзии, пароксизмальный амавроз, пароксизмальное сужение полей зрения с появлением скотом), глазодвигательные нарушения (трепетание век, нистагм, девиация глазных яблок в контралатеральную очагу сторону), вегетативные расстройства (головная боль, рвота, побледнение лица, двусторонний миоз) и ассоциативные феномены, связанные, с распространением возбуждения на теменную кору (анозогнозия, акалькулия, апраксия, аутотопагнозия). Типична высокая частота ЭП, особенно фокальных сенсорных зрительных приступов (стар.: изолированные зрительные ауры), иногда сливающихся в «фуги» или статус фокальных приступов высокой продолжительности (status amavroticus) [65]. Распространение возбуждения из затылочной коры вперед приводит к развитию лобной эпилепсии дополнительной моторной зоны [69]. В этих случаях начало приступа указывает на вовлечение зрительной коры.
Нередко у пациентов с СЗЭ выявляются косоглазие, нарушение конвергенции, снижение зрения, возможны зрительная агнозия («корковая слепота»), сужение полей зрения контралатерально очагу, когнитивные нарушения.
Иногда СЗЭ необходимо дифференцировать с мигренью и идиопатической фокальной (самоограничивающейся) затылочной эпилепсией, которые имеют сходство клинических проявлений.
На ЭЭГ обнаруживается региональная пик-волновая активность в одном из затылочных отведений, биокципитально, нередко с распространением на теменные и височные отведения или диффузно. В отличие от центротемпоральных спайков, выявляемых на ЭЭГ при идиопатической затылочной эпилепсии, эпилептиформная активность при СЗЭ не исчезает при записи ЭЭГ с открытыми глазами. При массивном структурном дефекте возможно появление продолженного регионального замедления [67]. Данные нейровизуализации зависят от этиологии СЗЭ. СЗЭ взрослых часто имеет серьезный прогноз, фармакорезистентность развивается у трети пациентов [71]. Дети с ФКД затылочной доли, как правило, – кандидаты на хирургическое лечение [72].
1.6.2.5 Синдром (энцефалит) Кожевникова – Расмуссена и эпилепсия Кожевникова
Эпилепсия Кожевникова (ЭК) - отдельная форма эпилепсии, но не нозологически самостоятельное заболевание, полиэтиологичное по своей природе, проявляющееся симптомокомплексом ЭК: облигатное наличие постоянного миоклонуса, обычно в сочетании с фокальными моторными, билатеральными судорожными эпилептическими приступами и очаговыми неврологическими симптомами [73]. Симптомокомплекс ЭК встречается при большом количестве различных неврологических заболеваний: энцефалиты (вирусные, энцефалит Кожевникова - Расмуссена, цистицеркоз и другие), инфекционные заболевания головного мозга с масс–эффектом (абсцесс, туберкулома, гумма), травматические, сосудистые (ишемические, геморрагические, венозный тромбоз), опухоли головного мозга, ФКД [74]. В России эпилепсия Кожевникова наиболее часто встречается при клещевом энцефалите и синдроме Кожевникова – Расмуссена.
Синдром (энцефалит) Кожевникова – Расмуссена (СКР) – тяжелое прогрессирующее заболевание головного мозга, подострый прогрессирующий очаговый энцефалит, предположительно аутоиммунной природы (не установлено). Возможные причины [75]: хроническая вирусная инфекция; острая вирусная инфекция, приводящая к локальным иммунным изменениям; аутоиммунный механизм, не связанный с инфекцией. У пациентов с СКР обнаружен повышенный титр антител к глутаматным (GluR3) рецепторам (не имеет диагностического значения) [76]. В соответствии с проектом классификации (2001) [56], СКР относится к группе симптоматической фокальной неокортикальной эпилепсии; по этиологической классификации эпилепсий (2017) [5] – к смешанной форме: иммунной и структурной.
Для СКР характерна клиническая триада: ЭП, двигательные нарушения (центральный гемипарез) и расстройство высших психических функций в сочетании с неуклонно прогрессирующим течением, тяжелой инвалидизацией и возможным летальным исходом. Дебют СКР в широком возрастном диапазоне, чаще – от 1 до 14 лет, с пиком в 5 - 8 лет, описаны случаи дебюта во взрослом возрасте [77]. Обычно заболевание начинается с фокальных моторных или билатеральных судорожных приступов, реже – фокальных приступов с автоматизмами и фокальных не моторных приступов, часто – с эпилептического статуса. Наиболее характерны ЭП, исходящие из моторной коры лобной доли. Выделяют 3 клинических стадии ЭКР [78]:
1 стадия (продромальный период, до 7 месяцев) – дебют заболевания с простых фокальных моторных ЭП (которым может предшествовать соматосенсорная аура) или с БТКП. Частота ЭП постепенно нарастающая. Постепенно формируется постприступный парез Тодда, присоединяются унилатеральные миоклонические приступы.
2 стадия (активный период, до 8 месяцев) - частые, продолжительные ЭП со статусным течением, постоянным эпилептическим миоклонусом в одной половине тела (симптомокомплекс ЭК). Постприступный парез Тодда сменяет перманентный гемипарез, присоединяются проводниковая гипестезия, выпадение полей зрения, расстройства психики и речи.
3 стадия (стабилизация, в 80% до 3 лет от начала заболевания) – стабилизация или урежение частоты ЭП при одновременном прогрессировании неврологических расстройств. У ¼ детей присоединяются нейроэндокринные нарушения: ожирение, преждевременное половое развитие [79].
Диагностические критерии СКР на основании European consensus statement [80] состоят из двух частей (А и В), представленных в таблице 3.
Таблица 3. Диагностические критерии СКР на основании European consensus statement
Часть А |
1. Клиника. Фокальные эпилептические приступы (с или без эпилепсии Кожевникова) в сочетании с односторонним кортикальным дефицитом. 2. ЭЭГ. Латерализованное по одной гемисфере замедление с эпилептиформной активностью или без нее. ЭЭГ- паттерны, указывающие на фокальный характер приступов. 3. МРТ. Фокальная кортикальная атрофия, локализованная в одной гемисфере, в сочетании хотя бы с одним из двух указанных ниже признаков: гиперинтенсивный сигнал от серого или белого вещества в режимах T2/FLAIR; гиперинтенсивный сигнал или атрофия ипсилатеральной головки хвостатого ядра. |
Часть В |
1. Клиника. Эпилепсия Кожевникова или нарастающий односторонний кортикальный дефицит. 2. МРТ. Нарастающая (при исследовании в динамике) фокальная кортикальная атрофия, локализованная в одной гемисфере. 3. Гистопатология (биопсия мозга). Критерий подтверждения: признаки энцефалита с преобладанием Т-клеток, активацией микроглии (формирование узелков в большинстве случаев, но не строго обязательно) и реактивным астроглиозом. Критерий исключения: большое количество паренхимальных макрофагов, В-клеток или плазменных клеток, а также включений частиц вирусов. |
Диагноз СКР может быть установлен только при наличии всех 3 критериев части А (первый этап) или любых 2 критериев части В (второй этап). Второй этап включает проведение церебральной биопсии. При установлении диагноза СКР важно обследование пациентов в динамике: сравнение клинических данных, результатов ЭЭГ и МРТ исследования. Прогноз при СКР всегда очень серьезный и определяется своевременностью хирургического лечения, которое позволяет остановить проявления разрушительной фармакорезистентной эпилепсии. Инвалидизация при СКР всегда выражена и обусловлена высокой частотой эпилептических приступов, наличием гемипареза и нарушением высших психических функций [76].
1.6.3. Возрастзависимые (синоним: генетические, самоограничивающиеся, самокупирующиеся) фокальные эпилепсии с началом в детском возрасте. Клиническая картина
Термин самоограничивающиеся эпилепсии означает возрастзависимый их характер с высокой вероятностью спонтанной ремиссии по приступам в определенном возрасте.
1.6.3.1. Возрастзависимая (самоограничивающаяся) эпилепсия с вегетативными приступами (ранее синдром Панайотопулоса)
Возрастзависимая (самоограничивающаяся) эпилепсия с вегетативными приступами – эпилептический синдром с началом в раннем детстве с фокальных вегетативных приступов, которые часто носят пролонгированный характер. Синдром относится к эпилепсиям со сложным типом наследования, оба пола страдают одинаково часто. Перинатальный анамнез не отягощен, у 5 - 17% детей в анамнезе фебрильные судороги.
Дебют приступов в возрасте от 1 года до 14 лет (чаще между 3 и 6 годом жизни). Приступы у большинства пациентов редкие, в 25% всех случаев имеет место единственный эпизод (он может быть эпилептическим вегетативным статусом), в 50% всех случаев общее число приступов за время болезни равно 6. У некоторых пациентов могут отмечаться довольно частые приступы. В 2/3 всех случаев приступы отмечаются во сне. Во время приступа характерны гастроинтестинальные симптомы (тошнота, рвота), бледность, цианоз, расширение зрачков, нарушения терморегуляции, частоты дыхания и сердцебиения. Могут отмечаться недержание мочи и гиперсаливация. По мере развития приступа возникает нарушение сознания, развивается поворот головы и глаз в сторону, иногда - гемиклонии. Приступ продолжается от нескольких минут до часов с формированием вегетативного эпилептического статуса. По завершении статуса резидуального неврологического или когнитивного дефицита нет.
Неврологический статус детей нормален, их развитие и когнитивные функции не страдают. Тем не менее в период активного течения эпилепсии может отмечаться негрубый речевой дефицит.
На межприступной ЭЭГ – на фоне нормальной биоэлектрической активности мультифокальные повторные спайки или комплексы острая-медленная волна высокой амплитуды у 90% детей, разряды могут менять свою локализацию. Наиболее частая локализация - затылочные отведения (в 60% всех случаев). В небольшом проценте случаев могут отмечаться генерализованные разряды и низкоамплитудные спайки. Разряды блокируется открытием глаз, так как имеет место fixation-off sensitivity (рефлекторный феномен ЭЭГ, вызванный устранением зрительной фиксации, которая определяется центральным зрением). Данный феномен чаще встречается при синдромах Панайотопулоса и Гасто, но может отмечаться и при идиопатических генерализованных эпилепсиях. В 10 % всех случаев однократная рутинная ЭЭГ разрядов не выявляет, что служит показанием к проведению ЭЭГ сна.
МРТ головного мозга в пределах нормы. Если у пациента отсутствуют атипичные черты синдрома, то проведение МРТ не является обязательным.
Спонтанная ремиссия наступает обычно через несколько лет с момент начала эпилепсии (в возрасте 11 - 13 лет).
1.6.3.2. Возрастзависимая (синоним - самоограничивающаяся) эпилепсия с центротемпоральными спайками (синоним - доброкачественная эпилепсия детства с центротемпоральными спайками или роландическая эпилепсия)
Возрастзависимая (синоним - самоограничивающаяся) эпилепсия с центротемпоральными спайками - эпилепсия раннего школьного возраста с короткими гемифациальными приступами, типичными разрядами на ЭЭГ и спонтанной ремиссией в позднем подростковом возрасте.
Имеет сходные клинические и энцефалографические черты с атипичной детской эпилепсией с центротемпоральными спайками, эпилептической энцефалопатией с продолженной спайк-волновой активностью во сне и синдромом Ландау – Клеффнера. Все эти синдромы могут рассматриваться как спектр состояний, и возможен переход пациента из одного синдрома в другой по мере течения болезни.
Тип наследования считается сложным (мультифакторным). Оба пола страдают одинаково часто. У сибсов могут отмечаться сходные разряды без приступов (аутосомно-доминантный тип наследования разрядов). В 10% всех случаев у родственников может отмечаться эпилепсия (чаще не детская эпилепсия с центротемпоральными спайками).
Перинатальный анамнез не отягощен. Возможны фебрильные судороги в анамнезе (в 5 - 15% всех случаев). В очень небольшом проценте случаев происходит эволюция из синдрома Панайотопулоса.
Начало приступов в возрасте от 3 до 14 лет (с пиком частоты в 9 - 10 лет). Приступ заключается в клонических сокращениях половины лица (губы, языка, щеки) и имеет лобно-теменно-оперкулярное происхождение. Отмечаются трудности артикуляции (афазия), жевания и глотания, а также гиперсаливация. Приступы короткие (менее 5 минут), редкие (большинство пациентов имеют менее 10 приступов за всю историю болезни), иногда бывает несколько приступов в течение дней или недель с последующим большим межприступным интервалом в несколько месяцев. Приступ может эволюционировать в ипсилатеральный клонический в руке, ипсилатеральный гемиклонический или в билатеральный тонико-клонический. Возможен парез Тодда.
Неврологический статус нормален. Развитие и когнитивные функции не страдают до начала эпилепсии. Во время ее активного течения может обнаруживаться поведенческий и когнитивный дефицит, особенно в речевой сфере. Если дефицит имеет серьезный характер, необходима запись ЭЭГ сна. Дефицит становится меньше, когда приступы проходят.
Межприступная ЭЭГ – нормальная биоэлектрическая активность с сохранением архитектуры сна. Высокоамплитудные центротемпоральные спайки или комплексы острая-медленная волна, активируемые во время дремоты и сна. Могут быть как уни-, так и билатеральными, могут фиксироваться и вне центротемпоральной области – в теменной, затылочной и лобной доле. Имеют типичную морфологию с максимальным негативным компонентом в центротемпоральной области (C3/C4 и T3/T4) и максимальным позитивным компонентом в лобных отведениях. Если имеет место стойкое фокальное замедление без центротемпоральных спайков или диффузное замедление, необходимо думать о других эпилептических синдромах.
МРТ головного мозга нормальна или имеются неспецифические изменения. Если отсутствуют атипичные клинические и энцефалографические черты, то проведение МРТ не является обязательным.
Приступы обычно проходят к 13 годам, изредка к 18 годам.
1.6.3.3. Детская затылочная эпилепсия со зрительными симптомами (ранее - детская затылочная эпилепсия тип Гасто)
Детская затылочная эпилепсия тип Гасто – самоограничивающаяся детская эпилепсия с началом в детском возрасте с хорошо контролируемыми приступами и ремиссией, наступающей через 2 - 4 года после начала приступов.
Тип наследования не известен (предполагается сложный/полигенный). Примерно в трети всех случаев имеется отягощенность по фебрильным судорогам и эпилепсии, а также сообщается о мигрени у родственников (9 - 16%). Оба пола страдают одинаково часто.
Приступы начинаются в возрасте от 15 месяцев до 19 лет с пиком в 8 - 9 лет. Характерны частые фокальные сенсорные зрительные приступы, с быстрым началом, короткие (чаще несколько секунд, у большей части до трех минут, редко – продолжительностью до 20 минут). Зрительные феномены представляют собой разноцветные маленькие круги, возникающие на периферии зрения и двигающиеся горизонтально, постепенно увеличиваясь в размерах, что может сопровождаться поворотом глаз или головы в ипсилатеральную фокусу сторону. Могут отмечаться другие приступные симптомы со стороны затылочной доли, включая слепоту, сложные зрительные галлюцинации, зрительные иллюзии (например, иллюзия движения глазных яблок), боль в орбитальной области.
Перинатальный анамнез не отягощен.
Неврологический статус нормален. Развитие ребенка и его когнитивные функции не страдают, лишь у некоторых пациентов описано незначительное нарушение когнитивных функций.
ЭЭГ – на межприступной ЭЭГ у большинства пациентов на фоне нормальной биоэлектрической активности отмечаются спайки или комплексы спайк-медленная волна в затылочных отведениях. Иногда подобные изменения определяются только во сне. Характерна фоточувствительность по типу «fixation-off» - в 20 - 90%, фотосенситивность - в 15%. В 20 % всех случаев имеет место сочетание спайк-волновой активности с центротемпоральными спайками или генерализованными спайк-волновыми разрядами. На ЭЭГ в начале приступа отмечается уменьшение обычной спайк-волновой активности с последующим внезапным развитием быстрых ритмов низкой амплитуды в затылочных отведениях. В то же время возможно и наличие медленной спайк-волновой активности в момент клоний глазных яблок или приступной слепоты.
МРТ головного мозга нормальна.
Ремиссия по приступам наступает у 50 - 60% пациентов через 2 - 4 года с момента начала эпилепсии.
1.6.4. Эпилептические синдромы детского возраста, генерализованные (клиническая картина)
1.6.4.1. Фебрильные приступы (ФП). Фебрильные приступы плюс
ФП – эпизоды эпилептических приступов, возникающих в младенческом или детском возрасте во время лихорадки, не связанной с инфекцией. Около 5% детей в общей популяции имеют в анамнезе ФП. Риск трансформации ФП в эпилепсию составляет 2 - 5%, повторяемость – до 40% [81].
При ФП обнаружены различные генные и хромосомные аномалии, в частности, изменения в хромосоме 2q23-24 [82]. Идентифицировано более 20 генов, мутации в которых приводят к возникновению трех групп генетических эпилепсий, дебютирующих с ФП, которые могут иметь как доброкачественное, так и прогрессирующее течение с присоединением очаговой неврологической симптоматики и интеллектуального дефицита. ФП могут наблюдаться у больных с хромосомной патологией, при некоторых наследственных нейродегенеративных болезнях и моногенных наследственных синдромах. Их следует дифференцировать с нейроинфекциями (менингиты, энцефалиты), а также с тяжелой миоклонической эпилепсией младенчества, при которой ФП долгое время - единственный тип ЭП.
Типичные (простые) ФП - это короткий генерализованный приступ продолжительностью < 15 мин (в большинстве случаев самопроизвольно прекращающийся в течение 2 – 3 мин), не повторяющийся в течение 24 ч, возникающий во время эпизода лихорадки, не вызванный острым заболеванием нервной системы, у ребенка в возрасте от 6 месяцев до 5 лет, без неврологического дефицита. Они дебютируют в возрасте от 6 мес до 5 лет (ср. 20 мес), проявляются ГТКП; реже (10%) – фокальными моторными ЭП с остановкой активности и автоматизмами и «височными синкопе». Их продолжительность обычно не превышает 5 минут. Риск возникновения при наличии ФП у одного из родителей - 20%, у обоих родителей – 55% [81].
Атипичные (сложные) ФП характеризуются следующими проявлениями [74]: возраст дебюта до 6 мес. или после 5 лет; фокальные проявления приступа (версия головы, гемиклонии), либо продолжительным течением более 15 мин, либо множественными последовательно повторяющимися приступами в рамках одного эпизода лихорадки.
При продолжительности приступа более 30 мин говорят о фебрильном эпилептическом статусе. Они имеют место в анамнезе 30% пациентов с резистентными формами эпилепсии; трансформируются в структурную фокальную эпилепсию, чаще в палеокортикальную височную (15% всех случаев).
Описан синдром идиопатической (генетической) генерализованной эпилепсии «фебрильные судороги плюс» [83], который наследуется по аутосомно-доминантному типу (локус 19q13.1; натриевая каналопатия), известны и другие его генетические варианты. При этом заболевании ФП возникают в сочетании с другими типами ЭП. Критерии диагноза: дебют от 4 месяцев до 9 лет; частые семейные случаи эпилепсии или ФП; наличие простых (типичных) ФП; облигатный тип приступов – частые генерализованные тонико-клонические судороги, возникающие как при температуре, так и без нее.
Показана возможность наличия при синдроме «фебрильные судороги плюс» не только генерализованных, но и фокальных эпилептических приступов, чаще по тип
· Рекомендуется установление диагноза «Эпилепсия» согласно следующим критериям: наличие двух неспровоцированных (или рефлекторных) эпилептических приступа, с интервалом > 24 ч или один неспровоцированный (или рефлекторный) приступ и вероятность повторения приступов, близкая к общему риску рецидива (≥ 60%) после двух спонтанных приступов в последующие 10 лет, или определить диагноз эпилептического синдрома (≥ 60% - следует трактовать как высокую вероятность рецидива) [3, 5, 133, 134].
Уровень убедительности рекомендаций C (уровень достоверности доказательств — 5).
· Рекомендуется всем пациентам с подозрением на эпилепсию подробный сбор жалоб и анамнеза с описанием приступов пациентом и очевидцами, анализ видеозаписи приступов для определения семиологии приступов [1, 2, 3, 133, 134].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5).
Комментарий: клиническая интерпретация пароксизмального события в пользу эпилептического приступа должна основываться на совокупности описания приступа, связанных с ним симптомов и дополнительной информации [135, 136]. Балльная оценка, основанная только на симптомах, позволила правильно классифицировать характер пароксизмов у 94% пациентов, диагностику эпилептических приступов с 94% чувствительностью и 94% специфичностью[137].
· Рекомендуется всем пациентам с подозрением на эпилепсию проведение дифференциального диагноза с другими неврологическими и соматическими заболеваниями с целью уменьшения риска диагностической ошибки и нанесения вреда пациенту по причине отложенного или неправильного лечения [138, 139, 460]. (План дифференциальной диагностики изложен в Приложении Б).
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 3).
Комментарий: Частота ложноположительных диагнозов эпилепсии варьирует от 2% до 71%. Ошибочный диагноз приводит к неправильному использованию ПЭП и влияет на возможность вождения автотранспорта и ограничивает трудоспособность пациентов[138 - 144].
· Рекомендуется консультация врача-кардиолога и комплексное кардиологическое обследование при подозрении на кардиогенную природу пароксизмальных расстройств [6, 460].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
· Рекомендуется всем пациентам с подозрением на эпилепсию подробный сбор жалоб и анамнеза на предмет наличия неспровоцированных (или рефлекторных) эпилептических приступов и дифференциального диагноза с психогенными неэпилептическими приступами [3, 145, 139, 460].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарий: Анамнез должен прояснить, что произошло с пациентом до, во время и после приступа. Важно описание пароксизмального события не только самим пациентом, но и свидетелем случившегося. Для разных типов эпилептических приступов характерен ряд клинических особенностей, поэтому диагностика должна основываться на совокупности клинических признаков. Подробный анамнез необходим для выявления наличия других типов эпилептических приступов: миоклонических, фокальных и абсансов, а также для исключения психогенных неэпилептических приступов (ПНЭП) [134, 460, 146, 147]. Дифференциально-диагностические критерии ПНЭП: продолжительность пароксизма более 10 минут [148], стереотипные движения тазом [149, 150], переменная амплитуда ритмичных движений [151, 152], сохранность сознания и памяти в момент приступа (чувствительность 63%, специфичность 96%) [153]. Постприступная спутанность сознания выявлена у 67% пациентов с эпилептическими приступами, в 100% случаев после ГТКП и у 13 - 16% пациентов с ПНЭП [149, 154].
· Рекомендуется всем пациентам с подозрением на эпилепсию собрать подробный наследственный анамнез для дифференциального диагноза с генетической формой эпилепсии [139, 146, 460].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарий: Риск любого типа эпилептических приступов у ребенка от матери с генетической генерализованной эпилепсией составляет 4 – 8%; от отца - риск незначительно выше, чем в общей популяции [146]. Если болен более, чем один родственник первой степени родства, то риск для ребенка будет 30% и более. Риск развития эпилепсии у сибсов пробанда с эпилепсией составляет 2,5 – 6,7%, а для детей пробанда - 1,6 – 6,3% [155].
· Рекомендуется всем пациентам с подозрением на эпилепсию консультация специалиста по диагностике и лечению эпилепсии для точной, ранней диагностики и своевременного лечения в соответствии с типом эпилептических приступов и формой эпилепсии и снижения риска преждевременной смерти [149, 150, 151, 157, 158].
Уровень убедительности рекомендаций С (уровень достоверности доказательств — 5).
Комментарий: Специалистом по эпилепсии является невролог, имеющий опыт диагностики, лечения и ведения пациентов с эпилепсией, что подтверждается соответствующим документом (удостоверением) о тематическом усовершенствовании в области эпилептологии (невролог-эпилептолог) [6]. Значительная часть диагнозов эпилепсии, установленных неспециалистами, оказывались неверными. Большинство ошибочных диагнозов было поставлено специалистами широкого профиля [140, 141].
· Рекомендуется всем пациентам с подозрением на эпилепсию проведение домашней видеозаписи пароксизмов, в том числе с использованием смартфона, для уточнения характера регистрируемых пароксизмов и проведения дифференциального диагноза с неэпилептическими приступами [1, 159].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 3).
Комментарий: Проспективное многоцентровое замаскированное клиническое исследование Tatum и соавт. [160] показало диагностическую точность интерпретации видео со смартфона при эпилептических приступах 89,1% для экспертов и 75,1% для резидентов. Chen и соавт.[152]сообщили о чувствительности 93% и специфичности 94% домашнего видео для дифференциации эпилептических приступов от психогенных не- эпилептических приступов. Beniczky и соавт. [161]сообщили об общей точности 85% в интерпретации семиологии приступов на основе видеорегистрации. Видеорегистрация пароксизмальных событий позволяет дифференцировать психогенные неэпилептические приступы от эпилептических и других физиологических событий с чувствительностью 95,4%, специфичностью 97,5% [162].
· Рекомендуется всем пациентам с подозрением на эпилепсию определить тип (ы) эпилептического приступа, типа эпилепсии и/или эпилептического синдрома, этиологии эпилепсии и коморбидных состояний на основании классификации эпилептических приступов МПЭЛ для дифференциального диагноза с неэпилептическими приступами и подбора противоэпилептической терапии [1, 5, 163].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
· Рекомендуется пациенту с подозрением на эпилепсию провести общий медицинский осмотр с оценкой состояния кожных покровов, сердечно-сосудистой системы, психического статуса, оценку психомоторного и речевого развития (у детей) для определения наличия/отсутствия клинического фенотипа генетической формы эпилепсии и дифференциального диагноза с другими неврологическими и соматическими заболеваниями [1, 133,164,165, 462].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
· Рекомендуется пациенту с впервые развившимся эпилептическим приступом проведение неврологического обследования для оценки неврологического статуса и определения риска рецидива эпилептических приступов [133, 463].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
Комментарий: P.R. Camfield и соавт. сообщили, что выявление очаговой неврологической симптоматики у детей после впервые развившегося эпилептического приступа позволило определить тип эпилептического приступа и прогнозировать рецидив в 30/168 случаях [166]. Риск рецидива приступов складывается из совокупности факторов риска: наличие наследственной отягощенности, перинатальные поражения мозга, перенесенные нейроинфекции и ЧМТ, наличие очаговой неврологической симптоматики, эпилептиформная активность на ЭЭГ, структурные изменения мозга, выявленные на МРТ. Риск рецидива приступов 60% и более соответствует высокому риску его повторения, и позволяет уже после первого эпилептического приступа диагностировать эпилепсию и назначать лечение [3, 5, 6].
· Рекомендуется детям и взрослым с впервые диагностированной эпилепсией проведение рутинного скрининга когнитивных и поведенческих нарушений с определением показаний для последующего специализированного нейропсихологического обследования [167,168].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарий: примерно у половины детей и взрослых когнитивные или поведенческие нарушения могут присутствовать уже в дебюте заболевания [169-171].
· Рекомендуется пациентам с эпилепсией при подозрении на когнитивный дефицит и поведенческие нарушения (по данным субъективных жалоб пациентов и их родственников на нарушение памяти, внимания или дезорганизацию в повседневной жизни) проведение специализированного нейропсихологического обследования (нейропсихологического тестирования) для объективизации когнитивных и поведенческих нарушений с целью последующего назначения терапии [166, 167].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарий: нарушения памяти, внимания, IQ, речевые и поведенческие нарушения выявляются как у пациентов с фокальной (височной) эпилепсией [174], так и у пациентов с идиопатическими (генетическими) генерализованными эпилепсиями, в частности, при юношеской миоклонической эпилепсии [172].
· Рекомендуется детям и взрослым с эпилепсией проведение специализированного нейропсихологического обследования в динамике (через 6-9 месяцев) для оценки эффективности лечения, выявления клинически значимых изменений когнитивных функций, поведения, связанных с приемом ПЭП, или после хирургического лечения эпилепсии [175, 176].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 3).
Комментарий: проведение нейропсихологического обследования в динамике поможет выявить нарушения памяти, речи или исполнительных функций, которые не позволяют пациенту следовать графику дозирования лекарств, предписанному врачом, побочные эффекты ПЭП [175, 176], снижение когнитивных функций на фоне повторных черепно-мозговых травм, эпилептического статуса или кластеров эпилептических приступов на фоне декомпенсированного течения эпилепсии [177-179].
· Рекомендуется пациентам с фармакорезистентной эпилепсией на этапе прехирургического обследования и в послеоперационном периоде проводить специализированное нейропсихологическое обследование для прогнозирования и выявления послеоперационного когнитивного дефицита [180 - 185].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 2).
Комментарий: после резекции височной доли, наиболее частого вида хирургического вмешательства, у 45% пациентов может наблюдаться ухудшение памяти [185, 186]. Операция может привести к ухудшению и развитию новых когнитивных нарушений [187]. Успешное хирургическое лечение эпилепсии у детей может положительно повлиять на последующее когнитивное развитие [188]. Аффективные нарушения наблюдаются почти у 50% кандидатов на хирургическое вмешательство [189], что требует дополнительной оценки и соответствующего лечения [190].
· Рекомендуется всем пациентам с подозрением на эпилепсию назначать следующие лабораторные анализы для проведения дифференциального диагноза и исключения сопутствующих заболеваний:
- Общий (клинический) анализ крови;
- Анализ крови биохимический общетерапевтический (исследование уровня общего белка, альбумина, мочевины, мочевой кислоты, креатинина, глюкозы, натрия, калия, кальция, магния, хлоридов, аспартатаминотрансферазы, аланинаминотрансферазы);
- По показаниям: количественное определение одной группы психоактивных веществ, в том числе наркотических средств и психотропных веществ, их метаболитов в крови иммунохимическим методом; количественное определение одной группы психоактивных веществ, в том числе наркотических средств и психотропных веществ, их метаболитов в слюне иммунохимическим методом; количественное определение одной группы психоактивных веществ, в том числе наркотических средств и психотропных веществ, их метаболитов в моче иммунохимическим методом.
Медицинское освидетельствование на состояние опьянения (алкогольного, наркотического или иного токсического)[191,192, 139, 463].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
Комментарий: данный перечень исследований рассматривается экспертами-членами рабочей группы как достаточный, но не исчерпывающий. План обследования должен исходить из клинической картины заболевания и других данных, полученных при объективных, лабораторных и инструментальных методах обследования.
· Рекомендуется пациентам с впервые развившимися эпилептическими приступами, лихорадкой, изменением психического статуса, менингеальными симптомами проведение люмбальной пункции и исследование ликвора с целью дифференциального диагноза с инфекционными, аутоиммунными заболеваниями [191, 193–196].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарий: необходимо провести исследование ликвора на цитоз, белок, содержание глюкозы (GLUT1).
· Рекомендуется пациентам с подозрением на эпилепсию иммунной этиологии с целью проведения дифференциального диагноза, определения типа антител и назначения иммунокорригирующей терапии назначать: анализ крови и цереброспинальной жидкости (ЦСЖ) на следующие антитела: NMDA-R, AMPA, glycine, GABAa-R, GABAb-R, DPPX, CASPR2, LGI1, IgLON5, GAD [195, 197-200, 464].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4).
· Рекомендуется пациентам с подозрением на аутоиммунную эпилепсию при наличии паранеопластического процесса с целью проведения дифференциального диагноза, определения типа антител и назначения иммунокорригирующей терапии назначать анализ на опухолевоспецифические антитела: LGI1 (тимома); ГАМК-В (мелкоклеточный рак легкого); ГАМК-А (тимома); NMDA-R (тератома яичника); GAD65(карцинома легкого); AMPA-R (тимома, мелкоклеточный рак легкого, аденокарцинома груди); mGluR5 (лимфома Ходжкина (50 – 70%)); ANNA-1 / Hu (мелкоклеточный рак легкого, нейроэндокринные опухоли); Ма-1 / Ма-2 (опухоль семенных клеток † , мелкоклеточный рак легкого); амфифизин (мелкоклеточный рак легкого, рак груди); ANNA-2 / Ri (мелкоклеточный рак легкого, рак груди); КАСПР-2 (тимома); глицин (тимома); DPPX (лимфома); GFAPα (тератома яичника); CRMP-5 (мелкоклеточный рак легкого, тимома); нейрексин-3α; МОГ; AK5; IgLON5 [197 - 201, 464].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4).
· Рекомендуется пациентам с подозрением на инфекционную эпилепсию при наличии клинико-инструментальной картины нейроцистицеркоза, с целью проведения дифференциального диагноза назначать анализ крови и ЦСЖ на антитела к T.solium[195, 202, 465].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
Комментарий: Наличие антител к T.solium или антиген T.solium являются основным клиническим критерием для подтверждения нейроцистицеркоза [202]. По уровню антител в сыворотке крови можно судить о поражение организма. Антитела в ЦСЖ позволяют оценить активность кист и указывают на поражение ЦНС. Лабораторные исследования малоинформативны при неактивных кальцифицированных кистах[203].
· Рекомендуется пациентам с подозрением на инфекционную эпилепсию при наличии ВИЧ-инфекции с целью проведения дифференциального диагноза и назначения специфической терапии назначать анализ крови на антитела классов M, G (IgM, IgG) к вирусу иммунодефицита человека ВИЧ-1, 2 (Human immunodeficiency virus HIV 1, 2) [204, 205, 466, 467].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4).
· Рекомендуется пациентам с подозрением на инфекционную эпилепсию при наличии клинико-инструментальных данных за нейротоксоплазмоз, с целью проведения дифференциального диагноза назначать молекулярно-биологическое исследование крови на токсоплазмы (Toxoplasma gondii), определение антител класса G (IgG) к токсоплазме (Toxoplasma gondii) в крови, определение антител класса M (IgM) к токсоплазме (Toxoplasma gondii) в крови, молекулярно-биологическое исследование ЦСЖ на токсоплазмы (Toxoplasma gondii), определение ДНК токсоплазмы (Toxoplasma gondii) в ЦСЖ методом ПЦР [206].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
· Рекомендуется пациентам с подозрением на инфекционную эпилепсию при наличии признаков цитомегаловирусной инфекции, с целью проведения дифференциального диагноза молекулярно-биологическое исследование ЦСЖ на цитомегаловирус (Cytomegalovirus) с применением метода ПЦР; молекулярно-биологическое исследование крови на цитомегаловирус (Cytomegalovirus) [207, 205].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
· Рекомендуется пациентам с подозрением на инфекционную эпилепсию, при наличии клинико-инструментальных данных за герпетический энцефалит, с целью проведения дифференциального диагноза определение антител к вирусу простого герпеса (Herpes simplex virus) в крови, определение антигенов вируса простого герпеса 1 и 2 типов (Herpes simplex virus types 1, 2) в крови, определение ДНК ВПГ 1 и 2 типа методом ПЦР в периферической крови и ликворе [208, 205].
Уровень убедительности рекомендаций C(уровень достоверности доказательств – 5).
Комментарий: по результатам исследования T. Armangueи и соавт. в 27% случаев после герпетического энцефалита формируется аутоиммунная эпилепсия [208].
· Рекомендуется пациентам с подозрением на инфекционную эпилепсию при наличии признаков вируса герпеса 6А/В, с целью проведения дифференциального диагноза назначать молекулярно-биологическое исследование периферической крови на вирус герпеса 6 типа (HHV6), определение ДНК вируса герпеса 6 типа (HHV6) методом ПЦР в периферической крови, качественное исследование, определение ДНК вируса герпеса 6 типа (HHV6) методом ПЦР в периферической крови, количественное исследование, определение антител к вирусу герпеса человека 6 типа (Herpes-virus 6) в крови, определение антител класса G (IgG) к вирусу герпеса человека 6 типа (Human herpes virus 6) в крови и ликворе [209, 205].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
· Рекомендуется пациентам с подозрением на инфекционную эпилепсию при наличии клинико-инструментальной картины клещевого энцефалита, с целью проведения дифференциального диагноза определять наличие антител класса M, G (IgM и IgG) к клещевому энцефалиту, ДНК возбудителя клещевого энцефалита в периферической крови и ликворе методом ПЦР [210, 211].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
· Рекомендуется пациентам с подозрением на инфекционную эпилепсию при наличии признаков Лайм-боррелиоза, с целью проведения дифференциального диагноза определять наличие антител класса M, G (IgM и IgG) к возбудителям иксодовых клещевых боррелиозов группы Borrelia burgdorferi sensu lato в ЦСЖ, ДНК возбудителей иксодовых клещевых боррелиозов группы Borrelia burgdorferi sensu lato в ЦСЖ методом ПЦР [212].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 4).
· Рекомендуется всем пациентам с подозрением на метаболическую этиологию эпилепсии назначать исследования, позволяющие определять биохимические нарушения: спектр аминокислот и ацилкарнитинов методом тандемной масс-спектрометрии; газовую хроматографию с масс-спектрометрией мочи; высокоэффективную жидкостную хроматографию и тандемную масс-спектрометрию органических кислот, определение активности биотинидазы; анализ спектра органических кислот мочи методом газовой хроматографии с масс-спектрометрией для проведения дифференциального диагноза и последующего назначения специфической терапии [213].
Уровень убедительности рекомендаций В (уровень достоверности доказательств –3).
Комментарий: Международная противоэпилептическая Лига признает восемь типов метаболической эпилепсии, а именно: дефицит биотинидазы и холокарбоксилазы синтазы, дефицит церебрального фолата, нарушения метаболизма креатина, судороги, реагирующие на фолиновую кислоту, дефицит переносчика глюкозы типа 1 (GLUT-1), митохондриальные и пероксисомные нарушения, пиридоксинзависимую эпилепсию. Однако выделяют и другие типы метаболических нарушений, связанных с развитием эпилепсии: нарушения цикла мочевины, глутаровая ацидурия, дефицит кофактора молибдена, некетотическая гипергликемия, некетотическая гиперглицинемия и дефицит янтарной полуальдегиддегидрогеназы [213].
· Рекомендуется пациентам с эпилепсией, принимающим противоэпилептические препараты (ПЭП), проведение терапевтического лекарственного мониторинга (ТЛМ) для индивидуализации лечения, коррекции дозы ПЭП с целью оптимизации лечения [213 - 218].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 4).
Комментарий: использование ТЛМ рекомендуется на стадии титрации и при изменении дозы ПЭП, после достижения желаемого клинического результата, с целью установления «индивидуального терапевтического диапазона» (два независимых измерения с интервалом 2 и 4 месяца); для определения величины увеличения дозы, особенно для ПЭП, показывающих дозозависимую фармакокинетику (особенно фенитоин**); при сомнениях в дифференциальной диагностике признаков или симптомов, указывающих на токсичность ПЭП, связанную с концентрацией, или когда токсичность трудно оценить клинически (например, у детей раннего возраста или у пациентов с психическими отклонениями); при продолжающихся эпилептических приступах, несмотря на очевидно адекватную дозу ПЭП. Проведение ТЛМ также необходимо при подозрении на изменение фармакокинетики из-за следующих факторов: переход в другую возрастную группу, беременность, сопутствующие заболевания или межлекарственные взаимодействия; для оценки потенциальных изменений в устойчивой концентрации ПЭП при изменении состава препарата, синонемической замене, в случае неожиданного изменения клинической реакции, при подозрении на нарушение пациентом режима лечения. К ПЭП с наивысшим уровнем рекомендации к проведению ТЛМ относятся: вальпроевая кислота** и карбамазепин**.
2.4.1. Электроэнцефалография
Введение: электроэнцефалография (ЭЭГ), (от слов электро – электричество; энцефалон – головной мозг; графия – пишу), является методом регистрации спонтанных колебаний тормозных и возбуждающих постсинаптических потенциалов, образующихся на дендритах пирамидных клеток коры головного мозга. ЭЭГ – основной метод функциональной диагностики центральной нервной системы у больных с эпилепсией. Различают: а) рутинную ЭЭГ – продолжительность 30 минут непрерывной записи; б) ЭЭГ сна – во время записи пациент спит, при этом на энцефалограмме регистрируют характерные паттерны, соответствующие стадиям сна; в) видео-ЭЭГ – сочетает не только регистрацию биоэлектрической активности головного мозга, но и синхронизированное по времени видеоизображение пациента. На практике возможны сочетания этих методик.
Методология анализа заключается в визуальном выделении на кривых особых графических элементов, специфичных эпилепсии (эпилептиформные графоэлементы) и их различных сочетаний. Различают интериктальную активность, наблюдаемую в межприступный период, и иктальную активность, которая характеризует эпилептический приступ. Биомаркером структурного повреждения головного мозга является фокальное замедление частоты биоэлектрической активности по отведениям, расположенным в соответствующей проекции. Для регистрации ЭЭГ следует руководствоваться рекомендациями экспертного совета по клинической нейрофизиологии Российской противоэпилептической лиги, а для описания использовать русскоязычный словарь терминов, используемых в клинической электроэнцефалографии [219, 220]. У пациентов с подозрением на эпилепсию или с уже установленным диагнозом ЭЭГ применяют после впервые возникшего эпилептического приступа для прогнозирования развития эпилепсии, для диагностики эпилептического синдрома, для оценки эффективности терапии.
· Рекомендуется проведение рутинной ЭЭГ с функциональными пробами пациентам, впервые в жизни перенесшим неспровоцированный эпилептический приступ для прогнозирования развития эпилепсии [220, 221, 468].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: выявление эпилептиформной активности на ЭЭГ после впервые возникшего эпилептического приступа свидетельствует о высоком риске возникновения повторного приступа. Чувствительность единичного исследования колеблется в диапазоне 20 - 55% и повышается по мере увеличения количества исследований до 90% [222 - 266].
· Рекомендуется проведение рутинной ЭЭГ с функциональными пробами пациентам с эпилепсией для диагностики эпилептического синдрома [227].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарий: В ряде случаев ЭЭГ помогает диагностировать соответствующий эпилептический синдром на основании выявления особых электрографических паттернов (таб. 8).
Таблица 8. Иктальные и интериктальные электрографические признаки эпилептических синдромов
Эпилептический синдром/группа синдромов |
Иктальная ЭЭГ |
Интериктальная ЭЭГ |
|---|---|---|
|
Абсансные эпилепсии с типичными абсансами |
3 Гц генерализованные комплексы спайк-волна, нередко повторяющиеся пробеги таких разрядов |
Нормальный фоновый ритм, активация интериктальных и иктальных разрядов при гипервентиляции |
|
Абсансные эпилепсии с атипичными абсансами |
1,5 – 2,5 Гц генерализованные разряды спайк-волна |
интериктальные разряды могут быть асимметричны с признаками фокальности |
|
Юношеская (синоним ювенильная) миоклоническая эпилепсия |
4 – 6 Гц генерализованная спайк- и полиспайк- волна |
Нормальный фон, часто активация интериктальных разрядов ритмичной фотостимуляцией; могут наблюдаться и более типичные 2.5 – 3 Гц разряды спайк-волна; интериктальные разряды могут быть асимметричны с признаками фокальности |
Инфантильные спазмы |
Генерализованная медленноволновая активность большой амплитуды и/или декремент активности |
В фоне полностью отсутствуют нормальные участки записи, доминирующим паттерном является гипсаритмия |
Синдром Ленокса - Гасто |
Зависит от типа приступа (атипичного абсанса, тонического или атонического приступа) |
А) медленные спайк-волновые разряды, активирующиеся во время сна и во сне больше, чем в бодрствовании, имеющие тенденцию к билатеральной синхронизации; Б) вспышки высокоамплитудных генерализованных полиспайков и комплексов полиспайк-медленная волна В) пробеги быстрой ритмичной активности с частотой 10 - 25 Гц, которые продолжаются несколько секунд (от 2 до 10) во время NREM сна |
Прогрессирующие миклонические эпилепсии |
Генерализованные и мультифокальные спайки, множественные спайки и острые волны |
Нарастающее замедление фона по мере прогрессирования болезни, в ряде случаев появление разрядов при ритмичной фотостимуляции |
|
Возрастзависимая (синоним самоограничивающаяся) фокальная эпилепсия детства с центротемпоральными спайками (ранее – доброкачественная эпилепсия детства с центротемпоральными спайками, синоним - Роландическая эпилепсия) |
Высокоамплитудные спайки или острые волны с максимумом над центрально-височной областью |
Нормальный фон; часто выраженное усиление разрядов во сне; разряды (высоко амплитудные центротемпоральные спайки или комплексы острая медленная волна) могут быть как билатеральные, так и с одной стороны, а тангенциальный диполь располагаться в продольном направлении спереди назад. Разряды могут отмечаться и вне центротемпоральной области |
Возрастзависимая (синоним самоограничивающаяся) эпилепсия с вегетативными приступами (синоним синдром Панайотопулоса, доброкачественная затылочная эпилепсия с ранним началом) |
Билатеральные или односторонние разряды спайк-медленная волна затылочной локализации |
Затылочные интериктальные разряды часто ослабляются при открытии глаз; фотостимуляция может привести к эпилептическому приступу |
Височная эпилепсия |
спайки, острые волны и перемежающаяся ритмичная дельта- активность в проекции височной доли, часто активирующиеся на фоне дремоты и сна |
Часто интермиттирующие или постоянное височное замедление; могут наблюдаться независимые интериктальные разряды с противоположной височной доли. |
Лобная эпилепсия |
Интериктальные разряды в лобной области |
Мезиальные лобные разряды которые часто невозможно зарегистрировать с помощью скальповой ЭЭГ, может наблюдаться вторичная билатеральная синхронизация |
· Рекомендуется у больных с подозрением на эпилепсию соблюдать непрерывную продолжительность записи рутинной ЭЭГ более 30 минут [228].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4).
Комментарий: продолжительность регистрации ЭЭГ прямо связана с частотой выявления эпилептиформных графоэлементов, однако однозначных рекомендаций о максимальной длительности исследования не существует [228].
· Рекомендуется регистрация ЭЭГ во время сна у детей с вновь возникшей или прогрессирующей задержкой психического развития для исключения синдрома продолженной (непрерывной) спайк-волновой активности во сне (Continuous spike and waves during sleep, CSWS) [229].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарий: Синдром эпилептической энцефалопатии развития и/или эпилептическая энцефалопатия с электрическим эпилептическим статусом медленного сна/активацией спайк-волновых разрядов во сне (ESES) ассоциирован с нейрокогнитивными нарушениями и редкими эпилептическими приступами у детей, его диагностика возможна только при регистрации ЭЭГ во время сна. Термин часто употребляют в качестве синонима электрического статуса медленного сна (ESES). По мнению нейрофизиологов, для описания находок на ЭЭГ следует использовать термин «Электрический эпилептический статус медленного сна». При индексе эпилептиформной активности ниже 85% и выше 50% решение о назначении противоэпилептической терапии следует принимать с учетом совокупности клинических данных и нейропсихологической оценки когнитивных функций в динамике.
· Рекомендуется регистрация ЭЭГ ребенку первых лет жизни с задержкой психоречевого развития [230].
Уровень убедительности рекомендаций С (уровень достоверности доказательств — 5).
· Рекомендуется проведение рутинной ЭЭГ бодрствования пациенту с подозрением на наличие детской абсансной эпилепсии с целью уточнения диагноза и в динамике для оценки эффективности терапии [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: У ребенка с детской абсансной эпилепсией, не получающего лечение, абсансы должны развиться сразу на правильно проведенной гипервентиляции. Если нет возможности сделать видеозапись приступа, лаборант должен использовать пробы со счетом вслух и внимательно наблюдать за клиническими проявлениями абсанса и оценить степень нарушения осознанности. Для оценки эффективности терапии целесообразно проводить рутинную ЭЭГ тогда, когда родители пациентов не видят приступов.
· Рекомендуется регистрация ЭЭГ для подтверждения не только клинической, но и электрографической ремиссии эпилепсии [231].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарий: преимущественно при генерализованных формах эпилепсии, количество эпилептиформной активности прямо коррелирует с вероятностью возникновения эпилептических приступов, поэтому решение о коррекции дозы противоэпилептических препаратов следует принимать после контроля ЭЭГ. Контрольную регистрацию ЭЭГ следует проводить с теми же условиями, что и первичную запись, выявившую эпилептиформную активность.
l Рекомендуется ЭЭГ сна или видео-ЭЭГ-мониторинг с регистрацией сна пациентам с подозрением на эпилепсию, у которых ЭЭГ бодрствования (короткая или длительная ) не информативна (не подтверждает наличие эпилепсии) [221, 232].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
l Рекомендуется регистрация ЭЭГ пациентам с эпилепсией не только для оценки электрографической ремиссии, но и для оценки эффективности лечения [221, 232].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
2.4.2. Длительный видео-ЭЭГ-мониторинг
Введение: Длительный видео-ЭЭГ-мониторинг – это синхронная регистрация ЭЭГ и видеозаписи состояния обследуемого. Основная цель видео-ЭЭГ-мониторинга – визуализация пароксизмальных событий для их детального анализа. Это важно для классификации эпилептических приступов, эпилептических синдромов и локализации эпилептогенного очага. Видео-ЭЭГ-мониторинг является важнейшим методом в дифференциальной диагностике эпилептических приступов и неэпилептических пароксизмальных расстройств, а также обязателен при подготовке к хирургическому лечению. Продолжительность обследования составляет от нескольких часов до нескольких дней. При проведении видео-ЭЭГ-мониторинга с целью подготовки к хирургическому лечению фармакорезистентной эпилепсии используют не только скальповые, чашечковые электроды, но, при необходимости, и инвазивные, которые имплантируют в полость черепа.
· Рекомендуется продолжительное, при необходимости - круглосуточное видео-ЭЭГ-мониторирование у пациентов с фармакорезистентной эпилепсией при подготовке к нейрохирургическому лечению [233, 234].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарий: Для определения показаний к хирургическому лечению и планированию операции по поводу фармакорезистентной эпилепсии обязательна синхронная регистрация ЭЭГ и видеозаписи семиологии нескольких эпилептических приступов для исключения их психогенного характера и локализации зоны начала приступа. Для этого используют многосуточное видео-ЭЭГ-мониторирование скальповыми или инвазивными электродами, продолжительность которого может достигать нескольких суток и даже недель.
· Рекомендуется при проведении скальпового видео-ЭЭГ-мониторирования у пациентов с фармакорезистентной эпилепсией во время подготовки к хирургическому лечению использовать фиксированные к скальпу чашечковые электроды, расположенные согласно международной схеме 10 - 20, а при подозрении на височную форму эпилепсии дополнительно устанавливать электроды в проекции нижних отделов височных долей по схеме 10 - 10 (нижняя височная цепочка) m [235].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарий: Продолжительный скальповый видео-ЭЭГ-мониторинг необходимо осуществлять чашечковыми электродами, прикрепленными к скальпу с помощью коллодия или медицинского клея, а при подозрении на височную форму эпилепсии число контактов, установленных на поверхность головы, может достигать 27. Это обусловливает требования к применяемым для этих целей энцефалографам: число регистрирующих каналов должно быть не менее 32, а в случае записей инвазивными электродами – 64.
· Рекомендуется проведение видео-ЭЭГ-мониторинга электродами, имплантированными в полость черепа (субдуральных, глубинных, овального отверстия, скуловых) пациентам с фармакорезистентной эпилепсией при невозможности локализовать зону начала приступа с помощью видео-ЭЭГ-мониторинга, выполненного скальповыми электродами [236].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарий: корковые электроды для регистрации ЭЭГ и электрокортикографии необходимо использовать в случаях, когда результаты скальповой записи не совпадают с данными МРТ и семиологией приступа, или их невозможно оценить из-за выраженных артефактов, чаще всего двигательных.
2.4.3. Показания для проведения видео-ЭЭГ-мониторинга у детей
l Рекомендуется запись видео-ЭЭГ-мониторинга по меньшей мере в течение 1 часа ребенку с подозрением на синдром Отахара с целью записи приступной ЭЭГ и для возможности оценки межприступной ЭЭГ как сна, так и бодрствования [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: Межприступный паттерн по типу «вспышка-подавление» при наличии эпилептических спазмов и тонических приступов (по отдельности или вместе) подтверждает диагноз. Возможны и фокальные приступы.
l Рекомендуется запись видео-ЭЭГ-мониторинга в динамике по меньшей мере в течение 1 часа ребенку с подозрением на синдром Отахара с целью оценки эффективности лечения [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
l Рекомендуется видео-ЭЭГ-мониторирование с записью ЭЭГ бодрствования и сна ребенку с подозрением на доброкачественные семейные, несемейные неонатальные и инфантильные судороги с целью оценки приступной и межприступной ЭЭГ[227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: Межприступная ЭЭГ нормальна или с незначительными эпилептиформными или неэпилептиформными фокальными или мультифокальными изменениями (Grintonetal., 2015). Изредка регистрируется паттерн «thetapointualternant», но он не специфичен для синдрома. Если удается записать приступ, то он начинается с диффузного билатерального уплощения активности в течение 5 - 20 секунд (соответствует тонической фазе приступа и/апноэ) с последующим развитием фокальных или билатеральных ритмичных высокоамплитудных волн, а затем с появлением острых волн в лобных, височных и центральных отведениях.
l Рекомендуется запись видео-ЭЭГ-мониторинга по меньшей мере в течение 1 часа, с включением сна, ребенку с фокальными приступами с ранним началом с целью записи приступной ЭЭГ и для возможности оценки межприступной ЭЭГ как сна, так и бодрствования [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
l Рекомендуется запись видео-ЭЭГ-мониторинга в динамике по меньшей мере в течение 1 часа, с включением сна, ребенку с фокальными приступами с ранним началом с целью оценки эффективности терапии [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
l Рекомендуется видео-ЭЭГ-мониторинг с включением сна и по крайней мере 10 минут после сна ребенку с подозрением на наличие инфантильных спазмов с целью подтверждения типа приступов и характеристики межприступной ЭЭГ [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: Инфантильные спазмы чаще наблюдаются при пробуждении ребенка. Гипсаритмия сначала возникает во сне, и только потом в бодрствовании, она исчезает в REM сон. У ребенка, как правило, отсутствуют физиологические паттерны сна (веретена, К-комплексы)
l Рекомендуется видео-ЭЭГ-мониторинг в динамике с включением сна и по крайней мере 20 - 30 минут после сна ребенку с диагнозом синдрома инфантильных спазмов (синоним - синдрома Веста) с целью оценки эффективности терапии [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
l Рекомендуется длительный видео-ЭЭГ-мониторинг с включением сна ребенку с подозрением на инфантильные спазмы при наличии разных типов приступов (не только спазмов) с целью подтверждения типа приступов, изменении характера приступов, или при отсутствии эффекта от терапии с целью уточнения диагноза [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
l Рекомендуется видео-ЭЭГ-мониторинг бодрствования и сна ребенку с подозрением на синдром Драве и Драве-подобные синдромы с целью уточнения диагноза [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: Диагноз синдрома Драве скорее основывается на клинических и генетических данных, данные ЭЭГ имеет дополнительное значение. У не получающего лечение пациента с синдромом Драве (атипичные фебрильные судороги и/или статусы) обнаружение замедления основной биоэлектрической активности (фокального или диффузного) с ранней фотосенситивностью и фиксацией приступов, свойственных синдрому Драве, подтверждает диагноз. Но четким подтверждением диагноза является обнаружение генетической природы заболевания.
l Рекомендуется видео-ЭЭГ-мониторинг бодрствования и сна в динамике ребенку с диагнозом синдрома Драве и Драве-подобного синдрома с целью оценки эффективности терапии [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
l Рекомендуется видео-ЭЭГ-мониторинг бодрствования и сна ребенку такой продолжительности, чтобы были зафиксированы приступы (желательно их серия) ребенку с подозрением на злокачественную эпилепсию детства с мигрирующими фокальными приступами с целью уточнения диагноза [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: Типичный «мигрирующий» приступный паттерн служит наряду с клиническими проявлениями важной составляющей синдрома. Приступный паттерн, локализованный в одной области и состоящий из ритмичной активности альфа-тета диапазона с ритмичными спайками и спайк-волнами, постепенно замедляется и останавливается. В этот момент он возникает в соседней области и прогрессивно по ней распространяется. При частых приступах приступный паттерн часто перемещается из одного полушария в другое.
l Рекомендуется видео-ЭЭГ-мониторинг бодрствования и сна ребенку с фебрильными судорогами плюс и с генетической эпилепсией фебрильными судорогами плюс с целью уточнения диагноза [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: Характерны нормальная биоэлектрическая активность, генерализованные спайк-волновые комплексы с частотой 2 - 3 Гц. Могут быть найдены другие типы приступов (абсансы, миоклонии, миоклонико-атонические, атонические). Может быть зарегистрировано начало фокального приступа (с лобных или с височных отведений).
l Рекомендуется видео-ЭЭГ-мониторинг бодрствования и сна ребенку с неклассифированной энцефалопатией развития и эпилептической для подтверждения диагноза [100, 227, 469].
Уровень убедительности рекомендаций C (уровень достоверности доказательств — 5).
l Рекомендуется видео-ЭЭГ-мониторинг с включением сна пациентам с возрастзависимой эпилепсией детства с центротемпоральными спайками (синоним роландической) в том случае, если рутинная ЭЭГ не информативна, с целью подтверждения диагноза [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
l Рекомендуется видео-ЭЭГ-мониторинг с включением сна пациентам с возрастзависимой эпилепсией детства с центротемпоральными спайками в том случае, если приступы частые, и есть подозрение на наличие структурной эпилепсии; при развитии атипичных абсансов и/или дроп атак и/или бессудорожного статуса, а также генерализованных тонико-клонических приступов бодрствования; при появлении серьезных трудностей обучения и речевых проблем с целью уточнения диагноза [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: На структурный характер этиологии эпилепсии может указывать изменение морфологии центротемпоральных спайков во сне, особенно развитие быстрых спайков или полиспайков, увеличение медленных волн, эпизоды депрессии ритма. Не характерны для данного синдрома значительное замедление основной биоэлектрической активности (фокальное или диффузное), пробеги диффузных билатеральных/генерализованных разрядов спайк-медленная волна длительностью более 3 секунд, а также отсутствие активации эпилептиформных разрядов во сне. Наличие продолженной спайк-волноволй активности во сне при наличии соответствующей клинической симптоматики (регресса психоречевого развития, частых фармакорезистентных приступов, приступов, не характерных для данного синдрома) может указывать на атипичную эволюцию синдрома.
l Рекомендуется видео-ЭЭГ-мониторинг с включением сна пациентам с возрастзависимой эпилепсией с вегетативными приступами (синдромом Панайотопулоса) в том случае, если рутинная ЭЭГ не информативна, с целью подтверждения диагноза[227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
l Рекомендуется видео-ЭЭГ-мониторинг (с записью электромиографии с дельтовидных мышц) с включением сна пациентам с подозрением на наличие миоклонико-атонической эпилепсии с целью подтверждения диагноза [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: У пациента, не получающего лечение, есть шанс зафиксировать миоклонии в первый час записи. Гипервентиляция может спровоцировать атипичные абсансы. Для фиксации атонического компонента миоклонически-атонического приступа пациент должен стоять или сидеть (под наблюдением, чтобы предотвратить травмоопасное падение).
l Рекомендуется видео-ЭЭГ-мониторинг с включением сна пациентам с возрастзависимыми эпилепсиями (с центротемпоральными спайками и с вегетативными симптомами) для оценки эффективности терапии и при решении вопроса о завершении лечения[227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: Наличие центротемпоральных разрядов не является поводом для продолжения терапии, они могут сохраняться длительно (до возраста 14 - 18 лет), не сопровождаясь эпилептическими приступами. Для решения вопроса об отмене терапии важна продолжительность клинической ремиссии.
l Рекомендуется видео-ЭЭГ-мониторинг (с записью электромиографии с дельтовидных мышц) с включением сна пациентам с подозрением на наличие синдрома Леннокса - Гасто с целью подтверждения диагноза[227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: Для регистрации тонических приступов, типичных для синдрома, иногда необходима запись нескольких часов сна.
l Рекомендуется видео-ЭЭГ-мониторинг (с записью электромиографии с дельтовидных мышц) пациенту с подозрением на или наличием миоклонуса (позитивного и негативного) и атонических приступов с целью подтверждения типа приступа [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
l Рекомендуется пролонгированный видео-ЭЭГ-мониторинг бодрствования (с записью электромиографии с дельтовидных мышц) пациентам с подозрением на наличие миоклонических абсансов с целью подтверждения диагноза [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: Абсансы провоцируются гипервентиляцией, поэтому эта провокация может проводиться повторно во время исследования. В положении сидя или лежа с закрытыми глазами пациент должен расслабиться, тогда миоклонии в руках будут более очевидны.
l Рекомендуется видео-ЭЭГ-мониторинг бодрствования и сна с подсчетом спайк-волнового индекса пациенту с подозрением на наличие энцефалопатии развития и/или эпилептической с активацией спайк-волновых разрядов во сне (син. эпилептической энцефалопатии с продолженной спайк-волновой активностью во сне/ электрическим эпилептическим статусом сна) или на наличие синдрома Ландау - Клеффнера с целью подтверждения диагноза [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: Желательна запись хотя бы одного цикла сна. Требуется определение типа и количества эпилептиформных изменений во время бодрствования и сна с подсчетом спайк-волнового индекса (процент продолжительности медленно-волнового сна, занятого разрядами). На современном уровне знаний трудно четко определить критическое значение спайк-волнового индекса (выше которого он считается патологическим). Большинство исследователей сходятся на том, что он должен быть 50% и выше. Тем не менее, нельзя трактовать спайк-волновый индекс в отрыве от клинических проявлений, так как иногда даже «массивная» эпилептиформная активность не влияет негативно на психоречевое развитие ребенка и не требует лечения (последняя ситуация требует нейропсихологического тестирования и динамического наблюдения).
l Рекомендуется видео-ЭЭГ-мониторинг бодрствования и сна с подсчетом спайк-волнового индекса в динамике пациенту с диагнозом энцефалопатии развития и/или эпилептической с активацией спайк-волновых разрядов во сне (синоним эпилептической энцефалопатии с продолженной спайк-волновой активностью во сне, электрическим эпилептическим статусом сна) или с синдромом Ландау - Клеффнера с целью оценки эффективности терапии[227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: Уменьшение спайк-волнового индекса сна (наряду с прекращением приступов и улучшением психоречевого развития) является одним из критериев эффективности терапии.
l Рекомендуется видео-ЭЭГ-мониторинг бодрствования пациенту с подозрением на наличие абсансов в тех случаях, если рутинная ЭЭГ не позволяет выявить абсанс, если на рутинной ЭЭГ есть четкое фокальное начало абсанса и персистирующие фокальные спайки в одной зоне, при отсутствии эффекта от проводимой терапии с целью уточнения диагноза [227, 469].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
2.4.4. Магнитно-резонансная томография
Введение: Магнитно-резонансная томография (МРТ) — метод лучевой диагностики, основанный на принципе ядерно-магнитного резонанса, возникающего при возбуждении протонов ядер водорода в исследуемой области после воздействия группы радиочастотных сигналов в сильном однородном магнитном поле. Преимуществами применения МРТ являются отсутствие ионизирующего излучения, произвольное направление срезов или сбор истинного трехмерного массива данных, а также получение морфологической (структурной), метаболической и функциональной информации об исследуемом органе либо системе.
Первоначальные протоколы МРТ для пациентов с эпилепсией включали, в дополнение к стандартному исследованию, импульсные последовательности толщиной среза 3 мм, с пространственным разрешением 0,7 х 0,7 — 0,8 х 0,8 мм, выполненные на аппаратах МРТ 1,5 Т, ориентированные параллельно и перпендикулярно оси гиппокампа. Такие протоколы давали дополнительную информацию о структурных изменениях височных долей, но значительно увеличивали время исследования с риском возникновения приступа и динамических артефактов во время проведения МРТ. Предусматривали также отдельные протоколы для пациентов с темпоральной, экстратемпоральной и другими видами эпилепсии [237]. Появление сверхвысокопольных МРТ и импульсных последовательностей с изотропным вокселем на аппаратах МРТ 1,5-3Т привели к созданию нового протокола структурной МРТ.
Высокое пространственное разрешение и контрастность изображения с охватом всего головного мозга при 3 Т МРТ напрямую связана с напряженностью магнитного поля. Тонкие структурные юкстакортикальные изменения, характерные для кортикальных дисплазий, склероза гиппокампа, аномалий развития головного мозга, требуют применения импульсных последовательностей минимальной толщиной среза (1-2мм и менее) с изотропным вокселем (то есть с элементами объемного образования в форме куба с равными гранями, снижающими эффект частичного объема). Согласно протоколу HARNESS-MRI (унифицированный протокол нейровизуализации с использованием МРТ при эпилепсии) структурная МРТ, независимо от марки аппарата МРТ, включает основные импульсные последовательности [238, 239] (таб. 9).
Таблица 9. Импульсные последовательности и характеристики МРТ при эпилепсии
Импульсная последовательность |
Толщина среза |
Размер вокселя, мм |
Плоскость сканирования |
Задача применения |
|---|---|---|---|---|
|
Т2-взвешенная (Т2 TSE) |
1 — 2 мм |
0,4 х 0,4 х 2 |
аксиальная, коронарная |
Оценка структуры гиппокампа |
|
Т1-взвешенные (Т1 GRE MPRAGE) |
1 мм и менее |
1,0 х 1,0 х 1,0 |
сагиттальная |
Оценка анатомии коры и белого вещества, диагностика дисплазий |
|
Т2 инверсия/восстановление с насыщением (отсечением) сигнала от свободной жидкости (Т2 TSE FLAIR/TIRM) |
1 мм |
1,0 х 1,0 х 1,0 |
сагиттальная |
Диагностика очаговых изменений, в т.ч. склероза гиппокампа |
МР-диффузия (DWI) |
3 мм |
1,5 x 1,5 x 3 |
аксиальная |
Дифференциальная диагностика ишемии, опухолей |
|
Импульсная последовательность, взвешенная по неоднородности магнитного поля (SWI/SWAN/Т2 Gre∗) |
1 — 2 мм |
1,0 х 1,0 х 1,0 |
аксиальная |
Диагностика кровоизлияний, сосудистых мальформаций, микрокровоизлияний в опухоль |
Данный протокол можно использовать на аппаратах МРТ 1,5 и 3 Т, с охватом всего головного мозга. Общее время сканирования при использовании многоканальных катушек (8, 12 или 32 канала) составляет не более 30 мин. Контрастное усиление при структурном МРТ показано только при подозрении на опухоль головного мозга, сосудистую мальформацию, инфекционный процесс. Противопоказания к МРТ стандартные и включают наличие несовместимого с магнитным полем кардиостимулятора, эндопротезов, стентов, портов для химиотерапии, беременность в первом триместре. Относительным противопоказанием является нарушение сердечного ритма. При исследовании пациентов в возрасте младше 5 лет может понадобиться анестезиологическое пособие для минимизации артефактов от движения во время МРТ.
У больных с подозрением на эпилепсию или с уже установленным диагнозом, структурную МРТ применяют после впервые возникшего эпилептического приступа для выявления источника судорожной активности, для дифференциальной диагностики, в динамике и для оценки эффективности хирургического лечения.
● Рекомендуется проведение структурной МРТ головного мозга (не менее 1,5 тесла) пациентам, впервые в жизни перенесшим неспровоцированный эпилептический приступ, для выявления возможной причины заболевания [238, 239].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
Комментарии: целью первоначального исследования является диагностика потенциальных источников судорожной активности: склероза гиппокампа, фокальной кортикальной дисплазии (ФКД), опухоли, постишемических и посттравматических изменений. Полученные данные необходимо коррелировать с выявленной эпилептиформной активностью на ЭЭГ. Следует помнить, что МР-негативная картина у пациента с впервые возникшим судорожным приступом не является доказательством отсутствия эпилепсии, диагноз устанавливается на основании комплексного анализа клинико-инструментальных данных.
● Рекомендуется повторение МРТ головного мозга на аппарате 3 Т по эпилептологическому протоколу пациентам с эпилепсией (по показаниям), независимо от результатов предыдущих исследований (исключение составляют пациенты с идиопатической генерализованной эпилепсией) [238 - 240].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
Комментарий: Обязательно проведение МРТ 3Т по эпилептологическому протоколу пациентам с фармакорезистентной эпилепсией для рассмотрения вопроса о хирургическом лечении. Отсутствие очаговых изменений при первичном МРТ, выполненном на аппарате МРТ 1,5Т по стандартной методике, не исключает диагноза эпилепсии; тонкие структурные юкстакортикальные изменения на аппарате МРТ 1,5Т не диагностируются в 50% случаев [241]. У 20 - 30% пациентов с височной эпилепсией и у 20 - 40% пациентов с экстратемпоральной эпилепсией при МРТ структурные изме
2.5.1. Диагностика моногенных вариантов идиопатических эпилепсий
Для установления этиологии моногенных вариантов эпилепсий используются несколько молекулярно-генетических методов: автоматическое секвенирование по Сенгеру, а также одновременный анализ мутаций в нескольких генах. Для диагностики заболеваний, характеризующихся специфическими клиническими признаками целесообразно проводить анализ мутаций в отдельных генах, ответственных за их возникновение. К таким заболеваниям относится туберозный склероз, нейрофиброматоз и синдром Ангельмана [21,22].
l Рекомендуется молекулярно-генетическое исследование мутаций в гене методом автоматического секвенирования по Сэнгеру пациенту с эпилепсией с подозрением на моногенное заболевание, для уточнения его этиологии и подбора специфической терапии [254, 255].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4).
Комментарии: Поиск мутаций в конкретном гене возможен только при хорошо фенотипически очерченных заболеваниях, ассоциированных с эпилепсией. Примером таких заболеваний могут служить синдром Ретта или туберозный склероз.
· Рекомендуется молекулярно-генетическое исследование мутации методом Сэнгера родителям пациента при выявлении у ребенка вероятно патогенной мутации или мутации с неясным клиническим значением в гене, ассоциированным с моногенным заболеванием, для уточнения значимости мутации в развитии эпилепсии и с целью прогноза деторождения [256].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
Комментарии: Отсутствие впервые выявленной нуклеотидной замены в гетерозиготном состоянии у здоровых родителей ребенка с заболеванием с аутосомно-доминантным типом наследования повышает вероятность ее патогенности. Обнаружение впервые выявленных нуклеотидных замен в гетерозиготном состоянии у родителей ребенка с двумя мутациями в гене, ответственном за возникновение аутосомно-рецессивного заболевания, повышает вероятность их патогенности и позволяет уточнить диагноз у пациента.
· Рекомендуется проведение исследования генов, ассоциированных с эпилепсией, путем высокопроизводительного секвенирования (NGS): панели генов, полноэкзомного или полногеномного секвенирования, пациенту с эпилепсией с подозрением на моногенную ее природу для установления причин генетически гетерогенной группы моногенных идиопатических эпилепсий [254].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 2).
Комментарии: Подозрение на моногенную природу эпилепсии возникает у пациента с отягощенным семейным анамнезом по эпилепсии, сочетанием эпилепсии с умственной отсталостью, расстройствами аутистического спектра и нарушениями движений, фармакорезистентностью эпилепсии и тяжелым ее течением (с эпилептическими статусами). Чаще всего необходимость такого исследования возникает при развитии ранней эпилептической энцефалопатии (синоним энцефалопатия развития и эпилептическая (см. раздел «Эпилептические энцефалопатии»).
Выраженная генетическая гетерогенность моногенных заболеваний и синдромов, сопровождающихся судорогами, а также значительный размер генов, ответственных за их возникновение обусловливает необходимость использование секвенирования нового поколения для диагностики отдельного генетического варианта этой группы заболеваний.
· Не рекомендуется проведение высокопроизводительного секвенирования в клинической практике пациентам с фокальными или генерализованными эпилепсиями с предположительно мультифакторным типом наследования с целью установления этиологии эпилепсии [257].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 2).
Комментарии: в редких случаях как при фокальных, так и при генерализованных эпилепсиях, особенно при наличии нескольких членов семьи с эпилепсией, может иметь место моногенная природа. В такой клинической ситуации генетическое исследование показано (например, при аутосомно-доминантной роландической эпилепсии или некоторых структурных формах эпилепсии, ассоциированные с нарушением нейрональной миграции или кортикальной организации).
· Рекомендуется цитогенетическое исследование (кариотип) пациенту с эпилепсией при подозрении на хромосомную природу эпилепсии с целью уточнения ее этиологии [258].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 3).
· Рекомендуется проведение хромосомного микроматричного анализа пациентам с эпилепсией, у которых предполагается хромосомный синдром, но при проведении стандартного кариотипирования количественных и структурных перестроек не выявлено [259].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 4).
Комментарии: Диагностика хромосомных синдромов, обусловленных микроаномалиями хромосом, размером менее 10 млн п.н. (делециями и дупликациями) проводится с использованием хромосомного микроматричного анализа, направленного на анализ вариаций числа копий ДНК у детей с эпилепсией с использованием SNP олигонуклеотидных микроматриц. Подозрение на хромосомную природу заболевания возникает в том случае, когда у пациента с эпилепсией, рожденного в срок с низким весом, ранее психомоторное развития протекало с задержкой, имеется множество стигм дисэмбриогенеза и/или пороки развития нескольких органов или систем организма. Некоторые виды хромосомной патологии могут быть диагностированы только методом кариотипа из свежей крови, такие как синдром кольцевой 20 хромосомы, характеризующийся сочетанием трудностей обучения с раннего возраста и бессудорожного эпилептического статуса и других типов эпилептических приступов.
· Рекомендуется полногеномное секвенирование пациенту с эпилепсией с подозрением на ее моногенную генетическую природу в том случае, если у него не выявлено причины эпилепсии при экзомном секвенировании с целью уточнения диагноза и подбора терапии [254].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 2).
Комментарии: если полногеномное секвенирование не проясняет молекулярно-генетический анализ, то такое же исследование показано и обоим родителям пациента.
· Рекомендуется пересмотр нативных данных экзомного или геномного секвенирования (представленных в формате fastQ) с предполагаемой генетической эпилепсией, если ранее не был установлен точный молекулярно-генетический диагноз [260].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4).
Комментарии: Необходимость такого подхода диктуется тем, что до сих пор остаются технические и биологические ограничения методов генетической диагностики, а также до сих пор не открыты значение и связь с фенотипом для всех генов. Пересмотр данных через полтора-два года является закономерным шагом в поиске и подтверждении генетической природы.
· Рекомендуется проведение анализа метилирования (метил-чувствительная ПЦР) СpG островков в области промотора гена SNRPN на хромосоме 15q11-q13 у ребенка с эпилепсией при подозрении на наличие у него синдрома Ангельмана с целью подтверждения диагноза [261].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии: Судороги - один их частых симптомов у пациентов с синдромов Ангельмана. Кроме эпилепсии у пациентов отмечается походка по типу «механической куклы», задержка речевого развития и характерная медленноволновая активность на межприступной ЭЭГ. Анализ метилирования позволяет выявить две основные причины возникновения синдрома-делеции в области хромосомы 15q11-q13 и однородительскую дисомию. В норме, у здоровых людей метилирован материнский аллель и не метилирован отцовский аллель в регионе 15q11-q13, что можно определить с помощью исследования промоторной области гена SNRPN (малого ядерного рибонуклеопротеина полипептида N) путем полимеразной цепной реакции (ПЦР). При синдроме Ангельмана анализ определяет неметилированный аллель отцовского происхождения при отсутствии метилированного материнского аллеля. Нормальный профиль метилирования не исключает синдром Ангельмана, потому что в 20 - 25% случаев этот синдром обусловлен мутацией гена UBE3A, и не сопровождается изменением метилирования.
2.5.2. Особенности клинических проявлений наследственных болезней обмена (НБО), сопровождающихся судорогами
Наследственные болезни обмена веществ, сопровождающиеся судорогами, –обширная группа моногенных заболеваний, обусловленных мутациями в генах, белковые продукты которых участвуют в сложных биохимических реакциях. Возникновение судорожного синдрома наблюдается в структуре симптомокомплексов более 200 нозологических форм, относящихся к различным классам НБО, в том числе аминоацидопатий, органических ацидурий, пероксисомных, митохондриальных и лизосомных болезнях накопления. Большинство заболеваний этой группы имеют аутосомно-рецессивный, Х-сцепленный рецессивный и митохондриальный тип наследования. Патогенез развития эпилепсии при этих группах нейрометаболических заболеваний, чаще всего, связан с дефицитом фермента, обусловленным мутациями в кодирующем его гене, что приводит к накоплениям метаболитов, предшествующих ферментативному блоку, оказывающих токсичное действие на клетки различных органов, вызывая их гибель. В связи с этим может поражаться как один «орган-мишень», так и несколько органов или систем, в том числе центральная и периферическая нервная система, скелетно-мышечная, сердце, печень, поджелудочная железа и даже кожа.
Особенностями клинических проявлений НБО, позволяющих заподозрить их наличие являются:
-необычный запах от кожи и ушной серы пациентов;
-острые метаболические кризы, которые часто протекают под маской нейроинфекции или провоцируются инфекционными заболеваниями;
-коматозные состояния;
-рвоты, непереносимость определенных продуктов.
Диагностика НБО
Диагностика наследственных нарушений обмена веществ может осуществляться на трех уровнях.
Первый уровень направлен на определение количества метаболитов нарушенных реакций. Он осуществляется с помощью тандемной масс-спектрометрии и высокожидкостной хроматографии органических кислот и газовой хроматографии/масс-спектрометрии мочи. С помощью этого метода диагностируются основные нозологические формы аминоацидопатий, органических ацидурий и болезней нарушения бета окисления жирных кислот. Достоинства заключаются в быстроте и дешевизне исследований, однако, маркеры могут обладать низкой специфичностью. Базовую информацию для дальнейшего диагностического поиска могут дать исследования кислотно-основного состава крови (включающим уровень рН), глюкозы, аммония, лактата, кетонов мочи и печеночного профиля. Одним из наиболее важных тестов является определение уровня сахара крови, так как гипогликемия является одним из частых симптомов НБО, сопровождающихся судорогами.
Магнитно-резонансная томография (МРТ) головного мозга может быть эффективна при диагностике НБО из группы лизосомных болезней накопления, пероксисомных болезнях, а также для обнаружения пороков развития мозга и его гипоксически-ишемических поражений.
Второй уровень – исследование активности определенного фермента или группы ферментов.
Третий уровень – ДНК диагностика, направленная на поиск мутаций в генах с помощью автоматического секвенирования по Сэнгеру, или использование метода высокопроизводительного секвенирования (NGS): панели генов, полноэкзомное и полногеномное секвенирование.
Учитывая возможность эффективной патогенетической терапии для некоторых нозологических форм этой группы заболеваний, их диагностику необходимо провести в кратчайшие сроки до появления необратимых повреждений головного мозга. Для нескольких нозологических форм эпилепсий из группы НБО, манифестирующих в неонатальном периоде разработана специфическая терапия, полностью купирующая клинические симптомы. К ним относятся четыре генетических варианта гипомагниемии, пиридоксин зависимые судороги и недостаточность биотинидазы. К возникновению гипомагниемии приводят мутации в четырех генах, продукты которых осуществляют абсорбцию магния в кишечнике или реабсорбцию магния в почечных канальцах, а к возникновению пиридоксинзависимых судорог – мутация в гене ALDH7A1, кодирующем фермент семейства альдегидфосфат дегидрогеназ. Недостаточность этого фермента приводит к накоплению пипередин-6 карбомилата, инактивирующего пиридоксальфосфат. Внутривенное введение электролитных растворов (препаратов магния) при наследственных гипомагниемиях или пиридоксальфосфата при пиридоксинзависимых судорогах полностью купирует симптомы и предотвращает их повторное возникновение. При отсутствии адекватного лечения формируется мышечная гипотония, возникает задержка темпов психомоторного развития и судорожный синдром. Эффективное лечение разработано и для наследственного дефицита биотинидазы - заболевания с аутосомно-рецессивным типом наследования.
· Рекомендовано комплексное определение концентрации на аминокислоты и ацилкарнитины в крови методом тандемной масс-спектрометрии, а также исследование аминокислот и метаболитов в моче пациенту с эпилепсией с подозрением на врожденный дефект метаболизма с целью уточнения этиологии эпилепсии и подбора терапии [262, 471].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5).
Комментарии Тандемная масс - спектрометрия является методом скрининга, направленного на анализ метаболитов, изменение концентрации которых обнаруживается при наследственных аминоацидопатиях, органических ацидуриях и нарушениях бета-окисления жирных кислот, в симптомокомплексе которых наблюдаются судороги.
Судороги являются одним из частых симптомов болезней обмена веществ. Одновременное определение нескольких десятков метаболитов в пятне крови позволяет провести анализ в кратчайшие сроки и спланировать алгоритм молекулярно-генетического исследования, направленного на уточнение нозологической формы и назначение своевременной терапии.
· Рекомендовано комплексное определение концентрации органических кислот в крови и комплексное определение содержания органических кислот в моче методом высокожидкостной хроматографии пациенту с эпилепсией с подозрением на врожденный дефект метаболизма с целью уточнения этиологии эпилепсии и подбора терапии [263].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
· Рекомендован комплекс исследований для диагностики недостаточности среднецепочечной ацил-КоА-дегидрогеназы жирных кислот и комплекс исследований для диагностики недостаточности длинноцепочечной 3-гидроксиацил-КоА-дегидрогеназы жирных кислот пациенту с эпилепсией с подозрением на врожденный дефект метаболизма из группы нарушения обмена жирных кислот и пероксисомных болезней, сопровождающихся судорогами, для уточнения этиологии эпилепсии и подбора терапии [264].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 3).
Комментарий: Определение метаболитов обмена жирных кислот, прежде всего фитановой кислоты позволяет заподозрить наличие пероксисомных заболеваний и спланировать алгоритм дальнейших поисков генетического варианта на основании использования секвенирования отдельного гена по Сенгеру.
· Рекомендуется исследование кислотно-основного состояния и газов крови ребенку с эпилепсией с подозрением на врожденный дефект метаболизма для поиска этиологии эпилепсии [265].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
· Рекомендуется исследование уровня молочной кислоты в крови пациенту с эпилепсией с подозрением на митохондриальную природу для уточнения этиологии эпилепсии и подбора терапии [266].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
Комментарии: существенное повышение лактата может свидетельствовать о митохондриальной природе эпилепсии.
· Рекомендуется исследование уровня пировиноградной кислоты в крови пациенту с эпилепсией с подозрением на ее митохондриальную природу для уточнения этиологии эпилепсии и подбора терапии [266].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
Комментарии: существенное повышение лактата и изменения соотношения лактата к пирувату может свидетельствовать о митохондриальной природе эпилепсии.
2.5.3. Особенности клинических проявлений хромосомных синдромов, сопровождающихся судорогами
К настоящему времени описано более 500 хромосомных аномалий, ассоциированных с нарушением ЭЭГ паттерна и судорогами. Дети с хромосомными синдромами часто рождаются с низким весом и множественными стигмами дизэбриогенеза и/или пороками развития двух и более органов или систем. В отличии от идиопатических моногенных эпилепсий у больных с хромосомными синдромами задержка развития, обычно, отмечается с рождения, и часто сопровождаются мышечной гипотонией. При некоторых хромосомных перестройках можно выделить характерный фенотип, например, при делеции короткого плеча 4 хромосомы (синдроме Вольфа - Хиршхорна), микроделеции короткого плеча 17 хромосомы (синдроме Миллера - Дикера), синдроме Ангельмана (микроделеция длинного плеча 15 хромосомы), но в большинстве случаев фенотип не специфичен. Определить хромосомную патологию можно цитогенетическим методом, но при малых размерах дисбаланса необходимо проведение молекулярного кариотипа (хромосомного микроматричного анализа). Лечение эпилепсии при хромосомной патологии симптоматическое.
2.5.4. Способы диагностики хромосомных синдромов, сопровождающихся судорогами
Диагностика количественных и структурных перестроек хромосом более, чем 10 млн п.н. осуществляется на основании традиционного метода анализа хромосом с использованием различных методов их окрашивания. Самым распространенным методом является G окрашивание по Романовскому - Гимзе. Использование этого метода позволяет выявлять полисомии, анеуплоидии, а также протяженные делеции, дупликации инверсии, инсерции, а также транслокации хромосом.
Диагностика структурных перестроек хромосом меньшего размера проводится с помощью хромосомного микроматричного анализа. При выполнении этого анализа исследуются все клинически значимые участки генома, позволяющие диагностировать все известные хромосомные синдромы (в том числе микроделеционные и микродупликационные).
Необходимо иметь в виду, что при проведении этого анализа не определяется наличие однородительских дисомий и транслокаций хромосом, а также перестройки за гранью разрешающей способности метода.
· Рекомендуется исследование кариотипа пациенту с эпилепсией при подозрении на наличие хромосомной патологии [258, 267]:
Уровень убедительности рекомендаций C (уровень достоверности доказательств — 3).
· Рекомендуется проведение хромосомного микроматричного анализа пациенту с эпилепсией при подозрении на хромосомную патологию при условии нормального кариотипа [259].
Уровень убедительности рекомендаций С (уровень достоверности доказательств - 4).
2.5.5. Диагностические мероприятия при оказании паллиативной помощи при эпилепсии у детей
· Рекомендуются первичные консультации врачей и иных специалистов из медицинской организации или ее структурного подразделения, оказывающего специализированную паллиативную медицинскую помощь детям с формами эпилепсии, относящимся к заболеваниям, ограничивающим продолжительность жизни (коды Международной классификации болезней Х пересмотра G 40.4, G 40.5, G 40.9) и детям с фармакорезистентной эпилепсией, которым выполнены паллиативные нейрохирургические вмешательства (каллозотомия и субпиальные транссекции), для определения объема необходимой паллиативной медицинской помощи [268- 272].
Уровень убедительности рекомендаций С (уровень достоверности доказательств - 5).
Комментарии: Термином «состояния, ограничивающие продолжительность жизни» (life-limiting conditions) у детей обозначаются состояния, при которых нет оправданной надежды на излечение и от которых дети умрут [268]. Детям с неизлечимыми заболеваниями или состояниями, сокращающими продолжительность жизни, оказывается паллиативная медицинская помощь [273]. Составленный в 2012 году на основании практического опыта пяти детских хосписов Великобритании «Словарь состояний детского возраста, ограничивающих продолжительность жизни» рубрицирован согласно разделам Международной классификации болезней 10 пересмотра (МКБ-Х) [268]. В частности, к состояниям, ограничивающим продолжительность жизни, данный словарь в блоке МКБ-10 «Эпизодические и пароксизмальные расстройства» относит коды G40.4 (Другие виды генерализованной эпилепсии и эпилептических синдромов) и G 40.5 (Особые эпилептические синдромы) [268]. Созданная в 2013 году «Директория состояний, ограничивающих продолжительность жизни» включает код МКБ-Х G40.9 (Эпилепсия неуточненная) [269]. Паллиативные нейрохирургические вмешательства у детей с фармакорезистентной эпилепсией (каллозотомия, субпиальные транссекции) [271, 272] требуют последующего оказания паллиативной медицинской помощи ребенку, что прямо следует из названия данной группы хирургических вмешательств.
· Рекомендуются повторные консультации врачей и иных специалистов из медицинской организации или ее структурного подразделения, оказывающего специализированную паллиативную медицинскую помощь детям с формами эпилепсии, относящимися к заболеваниям, ограничивающим продолжительность жизни (коды МКБ-ХG 40.4, G 40.5, G 40.9) и детям с фармакорезистентной эпилепсией, которым выполнены паллиативные нейрохирургические вмешательства (каллозотомия и субпиальные транссекции), для определения объема необходимой паллиативной медицинской помощи и оценки ее эффективности [268 - 272].
Уровень убедительности рекомендаций С (уровень достоверности доказательств - 5).
Обезболивание в лечении эпилепсии не применяется.
Показания и противопоказания к различным методам лечения – см. соответствующие разделы.
Медикаментозное лечение противоэпилептическими препаратами (ПЭП) является основным методом терапии эпилепсии. В большинстве случаев лечение должно начинаться сразу после установления диагноза «Эпилепсия» за исключением некоторых возрастзависимых синдромов детского возраста с редкими приступами. Цель лечения – достижение стойкой ремиссии заболевания без каких-либо значимых побочных эффектов (нервно-психических, соматических и др.); обеспечение профессиональной и социальной адаптации пациентов; сохранение оптимального качества жизни. В настоящее время в России зарегистрировано 23 ПЭП с разными механизмами действия, имеющие различный профиль эффективности и безопасности/переносимости.
Основные принципы терапии эпилепсии (выбор ПЭП в зависимости от типа приступов и формы эпилепсии/эпилептического синдрома, стартовая монотерапия, альтернативная монотерапия, рациональная политерапия, принципы комбинирования ПЭП и др.) являются общими для детей и взрослых, поэтому в основном рассматриваются в разделе «Медикаментозной терапии эпилепсии у взрослых». В разделе «Медикаментозная терапия эпилепсии у детей» рассматриваются возрастные аспекты противоэпилептической терапии и лечение специфических детских эпилептических синдромов.
Введение. При назначении ПЭП необходим персонализированный подход с учетом индивидуальных особенностей каждого пациента [6]. Выбор ПЭП зависит от формы эпилепсии/эпилептического синдрома, типа приступов, стадии и течения заболевания, пола, возраста, коморбидных состояний и других факторов.
Для правильного выбора ПЭП каждый врач в первую очередь должен установить является ли эпилепсия фокальной (с фокальными приступами с/без нарушения сознания, билатеральными тонико-клоническими с фокальным дебютом), генерализованной (с генерализованными приступами) или неуточненной (с недифференцированными приступами).
3.1.1.1. Лечение впервые диагностированной эпилепсии
Примерно 60 - 70% пациентов с эпилепсией достигают стойкой ремиссии или существенного урежения приступов на фоне лечения ПЭП. Большинство из них достигают ремиссии на монотерапии. Лечение начинают с начальной (стартовой) дозы наиболее подходящего ПЭП для данного пациента (персонализированный подход). Дозу постепенно увеличивают (титруют) до прекращения приступов или появления побочных эффектов. Каждый препарат имеет свою схему титрации.
3.1.1.1.1. Лечение впервые диагностированной фокальной эпилепсии (с фокальными приступами с/без нарушения сознания, билатеральными тонико-клоническими с фокальным дебютом)
В России при фокальных формах эпилепсии в режиме монотерапии разрешены к применению: бензобарбитал**, вальпроевая кислота**, габапентин, зонисамид, карбамазепин**, клоназепам**, лакосамид**, ламотриджин, леветирацетам**, окскарбазепин**, примидон, топирамат**, фенитоин**, фенобарбитал**, эсликарбазепин (перечень ПЭП здесь и далее указаны в алфавитном порядке).
· Рекомендуется начинать лечение взрослым пациентам с фокальной формой эпилепсии (фокальными эпилептическими приступами с/без нарушения сознания, билатеральными тонико-клоническими с фокальным дебютом) с вальпроевой кислоты**, габапентина, зонисамида, карбамазепина**, лакосамида**, ламотриджина, леветирацетама**, окскарбазепина**, примидона [274], топирамата**, фенитоина**, фенобарбитала**, эсликарбазепина в виде монотерапии с целью прекращения приступов [275–277].
Уровень убедительности рекомендаций А, (уровень достоверности доказательств – 1).
Для примидона: Уровень убедительности рекомендаций - В, (уровень достоверности доказательств – 2).
Комментарии: Необходимо отметить, что клиническая эффективность различных ПЭП у больных с фокальными формами эпилепсии при адекватных дозировках приблизительно одинакова и составляет 50 – 60 %.
В более ранних исследованиях при оценке эффективности базовых ПЭП было показано, что удержание на терапии у пациентов с фокальными приступами было значительно выше при назначении карбамазепина** и фенитоина** в сравнении с фенобарбиталом** и примидоном. При фокальных приступах с эволюцией в билатеральные тонико-клонические приступы удержание пациентов на терапии было значительно выше при лечении карбамазепином**, фенитоином** и фенобарбиталом** в сравнении с примидоном. Более ранняя отмена примидона и фенитоина** была связана с побочными эффектами. Таким образом, при фокальных приступах фенитоин**, примидон признаются так же эффективными, но не является препаратами первого выбора вследствие побочных эффектов. Клиническая эффективность бензобарбитала** в рандомизированных клинических исследованиях не изучалась.
Карбамазепин** долгие годы считался основным ПЭП для лечения пациентов с фокальными приступами; «золотым стандартом», с которым в рандомизированных клинических исследованиях сравнивали эффективность других ПЭП. Систематический обзор с сетевым метаанализом [275] показал, что длительность удержания на терапии (отражающая эффективность и переносимость) у пациентов с фокальными типами приступов, получающих ламотриджин и леветирацетам,** значительно выше в сравнении с карбамазепином**. В порядке убывания по сравнению с карбамазепином** по параметру удержания ПЭП распределились следующим образом: вальпроевая кислота**, зонисамид, окскарбазепин**, фенитоин**, топирамат**, габапентин, фенобарбитал**. Указанные ПЭП эффективны в лечении фокальных форм эпилепсии в режиме монотерапии.
Авторы другого метаанализа [276] рекомендуют начинать лечение с леветирацетама**, зонисамида, лакосамида**, эсликарбазепина в виде монотерапии при установлении диагноза фокальной формы эпилепсии (фокальных эпилептических приступах) у взрослых. Не было никаких статистически значимых различий в прекращении приступов через 6 и 12 месяцев при приеме указанных ПЭП. Все указанные ПЭП оказались эффективными и представляют собой подходящую альтернативу карбамазепину**.
Системный метаанализ[277], включающий 65 РКИ, в который вошел анализ использования леветирацетама**, ламотриджина, окскарбазепина**, топирамата**, вальпроевой кислоты** установил, что данные ПЭП являются эффективными для начальной монотерапии у взрослых. ПЭП не демонстрировали никаких признаков превосходства или неполноценности по сравнению с карбамазепином**, который считается стандартным лечением фокальной эпилепсии.
В рандомизированном клиническом исследовании [278] показано, что лакосамид** сравним по клинической эффективности с карбамазепином** у больных с впервые диагностированной эпилепсией.
Согласно результатам систематического обзора [279], нет однозначных доказательств, на основании которых возможен выбор между карбамазепином** и вальпроевой кислотой**. В работе взяты такие критерии как «время до отмены ПЭП» (удержание на терапии), «ремиссия на протяжении 12-месяцев», «время до первого приступа». По результатам исследования подтверждается рекомендация использования карбамазепина** в качестве ПЭП первого выбора у пациентов с фокальной эпилепсией.
При сравнительном анализе карбамазепина** и топирамата** [280] были получены данные, что карбамазепин** с меньшей вероятностью будет отменен и что 12-месячная ремиссия будет достигнута раньше, чем при применении топирамата**.
Систематический обзор 13 РКИ [281], сравнивающих карбамазепин** и ламотриджин, показал, что оба ПЭП в качестве монотерапии являются эффективными в лечении фокальных форм эпилепсии. Время прекращения приема («удержания») раньше наступала при монотерапии карбамазепином**. Наиболее распространенная причина прекращение приема – побочные эффекты: в 51% у карбамазепина** от общего числа случаев отмены против 36% у ламотриджина. Второй причиной отмены препарата был рецидив приступов на фоне лечения: 8% пациентов принимающих карбамазепин** и 15% ламотриджин. Полученные результаты также свидетельствуют о том, что рецидив приступов после начала лечения ламотриджином может произойти раньше, чем при лечении карбамазепином**, а ремиссия приступов в течении 6 месяцев может произойти раньше на карбамазепин**, чем на ламотриджин.
В проспективном двойном-слепом рандомизированном исследовании [282] был показан эквивалентный эффект леветирацетама** и карбамазепина** для достижения ремиссии приступов при впервые диагностированной фокальной эпилепсии.
Один из выводов научной группы исследования «КОМЕТ» гласит, что терапия леветирацетамом** не превосходит лечение карбамазепином** и ламотриджином в стандартных дозах у пациентов с впервые диагностированной эпилепсией [283].
В проведенном исследовании стандартных и новых ПЭП (Standart And New Antiepileptic Drugs, SANAD) с участием пациентов с фокальной эпилепсией (n = 1721) сравнивалась эффективность карбамазепина**, габапентина, ламотриджина, окскарбазепина**, топирамата** (проспективное, в параллельных группах, открытое, рандомизированное исследование в условиях реальной клинической практике в течение 12 мес., с точки зрения времени до констатации отсутствие эффекта терапии) [284]. Ламотриджин по клинической эффективности значительно превосходил карбамазепин** и окскарбазепин**, которые в свою очередь значительно превосходили габапентин и топирамат**. Эти различия в эффективности сохранялись в течение 6 лет. С точки зрения переносимости ламотриджин и габапентин значительно превосходили окскарбазепин**, который значительно превосходил карбамазепин** и топирамат**.
Интересным является научный подход оценки эффективности ПЭП при различных типах приступов. В 2013 г. экспертная группа МПЭЛ выпустила обзор эффективности начальной монотерапии у пациентов с недавно диагностированными или нелеченными различными эпилептическими приступами и двумя эпилептическими синдромами [285]. Результаты были основаны на 64 РКИ и 11 метаанализах, завершенных за последние 72 года. Авторы пришли к следующим выводам по уровням доказательности для взрослой когорты пациентов: взрослые с фокальными приступами – эффективность установлена: карбамазепин**, леветирацетам**, фенитоин**, зонисамид; вероятно эффективен: вальпроевая кислота**; возможно эффективен: габапентин, ламотриджин, окскарбазепин**, фенобарбитал**, топирамат**; потенциально эффективен: клоназепам**, примидон; пожилые люди с фокальными приступами – эффективность установлена: габапентин, ламотриджин; вероятно эффективен: нет ПЭП; возможно эффективен: карбамазепин**; потенциально эффективен: топирамат**, вальпроевая кислота**.
Авторы системного обзора, оценивавших эффективность клоназепама**, пришли к выводу об отсутствии доказательств для рекомендации данного ПЭП взрослым пациентам с фокальными формами эпилепсии в виде монотерапии [286].
· Рекомендуется начинать лечение у пожилых пациентов (старше 65 лет) с фокальной формой эпилепсии (фокальными эпилептическими приступами с/без нарушения сознания, с билатеральными тонико-клоническими с фокальным дебютом) с леветирацетама**, ламотриджина в виде монотерапии с целью прекращения приступов [287].
Уровень убедительности рекомендаций А, (уровень достоверности доказательств – 1).
Комментарии: Показана лучшая переносимость (меньшая частота побочных эффектов) у леветирацетама**, ламотриджина, габапентина в сравнении с карбамазепином** у пожилых пациентов [287, 288]. Пожилым пациентам с учетом снижения функции почек, накоплении коморбидных заболеваний, сопутствующей терапии рекомендуется начинать терапию с более низких доз ПЭП, выбирать более низкий темп титрации. Необходимо также учитывать возможное фармакокинетическое взаимодействие ПЭП с другими препаратами, получаемыми пациентом.
Ламотриджин и леветирацетам** являются более предпочтительными по сравнению с карбамазепином** у пожилых пациентов с фокальными формами эпилепсии по причине их лучшей переносимости и небольшой вероятности фармакокинетического взаимодействия с другими препаратами [287].
· Рекомендуется начинать лечение у пожилых пациентов (старше 60 лет) с фокальной формой эпилепсии (фокальными эпилептическими приступами с/без нарушения сознания, с билатеральными тонико-клоническими с фокальным дебютом) с габапентина в виде монотерапии с целью прекращения приступов [288].
Уровень убедительности рекомендаций А, (уровень достоверности доказательств – 2).
Комментарии: Габапентин по эффективности сравним с карбамазепином** и лучше переносится у пациентов старше 60 лет с впервые диагностированной фокальной эпилепсией. Применение габапентина приводит к урежению частоты приступов у пациентов старше 60 лет [288].
Выбрав препарат, врач должен назначить индивидуальную эффективную дозу. Доза ПЭП постепенно повышается (согласно схеме титрации) до достижения ремиссии приступов или появления побочных эффектов. Предпочтительны ретардные формы ПЭП, которые можно принимать 1 - 2 раза в сутки, что повышает комплаентность пациента к лечению. Как правило, это ПЭП нового и новейшего поколения.
Только после достижения максимально переносимой дозы и отсутствия у пациента ремиссии приступов можно говорить о неэффективности ПЭП и планировать замену на ПЭП следующей очереди выбора в виде вторичной монотерапии либо перейти к рациональной политерапии. Знание механизмов действия ПЭП приобретает наибольшее значение при выборе препаратов для комбинированной терапии. В настоящее время у экспертов нет однозначного ответа на вопрос «комбинировать или заменять» после первой неудачной монотерапии. В целом принято считать, что замена ПЭП более целесообразна при плохой переносимости и малой эффективности первого препарата, в то время как рациональная политерапия возможна в случае хорошей переносимости первого ПЭП, но недостаточной его эффективности.
Известные механизмы действия, начальные и среднетерапевтические дозы ПЭП, схемы титрации в сокращенном виде указаны в приложении №А3.
3.1.1.1.2. Лечение впервые диагностированной генерализованной эпилепсии (генерализованных приступов)
В России при генерализованной эпилепсии в режиме монотерапии разрешены к применению: бензобарбитал**, вальпроевая кислота**, карбамазепин**, клоназепам**, ламотриджин, окскарбазепин**, примидон, топирамат**, фенитоин**, фенобарбитал**.
Наиболее часто генерализованные формы эпилепсии дебютирует в детском и подростковом возрасте. «Тезисы-рекомендации» по лечению данных форм будут представлены в соответствующем разделе.
· Рекомендуется начинать лечение у взрослых пациентов с генерализованными формами эпилепсии (c генерализованными тонико-клоническими приступами) с вальпроевой кислоты**, карбамазепина**, ламотриджина, окскарбазепина**, топирамата**, фенитоина**, фенобарбитала** в виде монотерапии с целью прекращения приступов [275].
Уровень убедительности рекомендаций А, (уровень достоверности доказательств – 1).
Комментарии: Длительное время препаратом выбора при генерализованных формах эпилепсии (генерализованных приступах) считались препараты вальпроевой кислоты**. Высококачественные данные обзора [275] рандомизированных контролируемых исследований монотерапии у взрослых с генерализованным началом тонико-клонических приступов (с другими генерализованными типами приступов или без них) подтверждают использование вальпроевой кислоты** в качестве первой линии лечения, но при этом демонстрируют, что ламотриджин и леветирацетам** (согласно инструкции является средством дополнительной терапии при генерализованных приступах) могут быть подходящей альтернативой вальпроевой кислоте**. Это особенно важно для женщин с детородным потенциалом, для которых вальпроевая кислота** не является препаратом выбора из-за ее высокой тератогенности.
Ламотриджин имеет сравнимую клиническую эффективность с карбамазепином** в отношении первично-генерализованных тонико-клонических приступов [282].
В РКИ вальпроевая кислота** лучше переносилась, чем топирамат**, и была более эффективной, чем ламотриджин. Авторами сделан вывод, что вальпроевая кислота** должна оставаться препаратом первого выбора для многих пациентов (прежде всего, лиц мужского пола) с генерализованными эпилепсиями. Однако из-за потенциальных побочных эффектов вальпроевой кислоты** ее использование следует ограничить у девочек и женщин репродуктивного возраста, за исключением тех случаев, когда другие методы не эффективны или противопоказаны [284, 289].
В системном обзоре, исследующим эффективность монотерапии топираматом** у пациентов с юношеской миоклонической эпилепсией, не было существенных различий в эффективности между топираматом** и вальпроевой кислотой** у пациентов, ответивших на лечение снижением количества миоклонических или первично-генерализованных тонико-клонических приступов на 50% и более [290].
Мнение о том, что вальпроевая кислота** превосходит карбамазепин** при генерализованных тонико-клонических приступах в структуре генерализованной эпилепсии, не подтверждается этими данными [279].
Согласно системному обзору МПЭЛ (64 РКИ и 11 мета-анализов) имеется следующая доказательная эффективность начальной монотерапии у пациентов с генерализованными приступами: у взрослых с генерализованным началом тонико-клонических приступов – доказанная эффективность: нет ПЭП; вероятная эффективность: нет ПЭП; возможная эффективность: карбамазепин**, ламотриджин, окскарбазепин**, фенобарбитал**, фенитоин**, топирамат**, вальпроевая кислота**; потенциальная эффективность: габапентин, леветирацетам**; для пациентов с юношеской миоклонической эпилепсией: доказанная эффективность – нет ПЭП, вероятная эффективность – нет ПЭП, возможная эффективность – нет ПЭП, потенциальная эффективность – топирамат**, вальпроевая кислота** [285].В этом же обзоре указано, что согласно нерандомизированным клиническим исследованиям – карбамазепин**, габапентин, окскарбазепин**, фенитоин** могут аггравировать абсансные и миоклонические приступы, а в некоторых случаях и генерализованные тонико-клонические приступы. Имеются данные о том, что ламотриджин может аггравировать миоклонические приступы при ЮМЭ.
Клинических исследований, оценивающих эффективность/переносимость клоназепама**, при генерализованных эпилепсиях у взрослых пациентов в виде монотерапии нет.
3.1.1.2. Лечение фармакорезистентной эпилепсии
Введение. Фармакорезистентная эпилепсия (ФРЭ) – форма заболевания, при которой приступы продолжаются, несмотря на адекватную противоэпилептическую терапию двумя ПЭП в виде монотерапии или в комбинации. Доля больных с ФРЭ варьирует от 20 до 30%. Перед коррекцией лечения необходимо убедиться в отсутствии псевдорезистентности, которая чаще всего связана с неправильным диагнозом, неправильно выбранным ПЭП или его дозой, плохой комплаентностью пациента к лечению.
Только при недостаточной эффективности правильно подобранной монотерапии возможна политерапия. Как правило, политерапия целесообразна после не менее чем двух последовательных попыток применения препаратов в режиме монотерапии.
В соответствии с общепринятым мнением, при проведении политерапии следует комбинировать ПЭП с различными механизмами действия, чтобы повысить эффективность и избежать увеличения риска возникновения нежелательных явлений. В связи с большим выбором ПЭП при переходе на политерапию необходимо учитывать фармакокинетические и фармакодинамические особенности назначаемых ПЭП, т.е. речь идет о рациональной политерапии.
· Рекомендуется направлять взрослых пациентов с установленной фармакорезистентностью (ФР) в эпилептологический центр или в специализированный нейрохирургический центр для консультации врача-нейрохирурга с целью решения вопроса о возможности нейрохирургического лечения эпилепсии [373].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 3).
l Рекомендуется назначение клобазама в качестве дополнительной терапии взрослым пациентам с эпилептическими приступами любого типа при фармакорезистентной эпилепсии с целью лечения эпилепсии [330].
Уровень убедительности рекомендаций А (уровень достоверности доказательств – 1).
Комментарии: клобазам зарегистрирован в РФ в 2020 г. Является препаратом третьей очереди выбора в лечении эпилепсии (преимущественно фармакорезистентной).
Лучше переносится, чем клоназепам** (меньше седативный эффект), к нему реже, чем к клоназепаму**, развивается привыкание с истощением эффекта. Отмена препарата, также как клоназепама**, должна быть медленной, чтобы избежать синдрома отмены.
●Рекомендуется введение защечного раствора ♯мидазолама в дозе 10 мг взрослым пациентам при возникновении кризисной ситуации с развитием судорог продолжительностью более 5 минут с целью их купирования и профилактики эпилептического статуса [434, 547].
Уровень убедительности рекомендаций C (уровень достоверности доказательств 5).
●Рекомендуется введение ректального раствора ♯диазепама в дозе 10 мг взрослым пациентам при возникновении кризисной ситуации с развитием судорог продолжительностью более 5 минут с целью их купирования и профилактики эпилептического статуса [434].
Уровень убедительности рекомендаций C (уровень достоверности доказательств 5).
Комментарии: ректальный раствор #диазепама и защечный раствор #мидазолама включены ВОЗ в типовой список основных лекарственных средств на 2021г, рекомендованный всем странам иметь как минимальный в наборе жизненно важных лекарств у детей и взрослых (WHO Model List of Essential Medicines – 22nd List 2021). Очень часто данные лекарственные препараты назначаются пациентам с эпилепсией, которые признаны нуждающимися в паллиативной медицинской помощи. Дети, получающие паллиативную медицинскую помощь, которым назначены диазепам ректальный и мидазолам защечный, должны иметь возможность продолжить подобранную терапию и после достижения возраста 18 лет, т.е. должна соблюдаться преемственность между детской и взрослой эпилептологической службой.
3.1.1.2.1. Лечение фармакорезистентной фокальной эпилепсии (с фокальными приступами с/без нарушения сознания, билатеральными тонико-клоническими с фокальным дебютом)
· Рекомендуется назначать в качестве дополнительного средства у взрослых пациентов с фармакорезистентной фокальной эпилепсией (фокальными эпилептическими приступами с/без нарушения сознания, с билатеральными тонико-клоническими с фокальным дебютом): бриварацетам**, вальпроевую кислоту**, габапентин, зонисамид, лакосамид**, ламотриджин, леветирацетам**, окскарбазепин**, перампанел, прегабалин**, топирамат**, с целью прекращения приступов [292, 293, 294, 295, 528].
Уровень убедительности рекомендаций А (уровень достоверности доказательств – 1).
Эсликарбазепин [295].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 2).
Комментарии: По результатам большого системного обзора с мета-анализом [292] для выявления и сравнения профиля эффективности/переносимости 11 ПЭП показано, что все препараты были эффективнее, чем плацебо, в качестве дополнительного ПЭП к базовой терапии с редукцией приступов более 50%. Метаанализ показал, что леветирацетам**, вальпроевая кислота**, габапентин имеет лучшее сочетание по краткосрочной эффективности и переносимости. Окскарбазепин** хотя и был столь же эффективен, переносился хуже, чем эти три ПЭП. Остальные ПЭП - топирамат**, зонисамид, прегабалин**, ламотриджин, лакосамид** демонстрировали меньшую кратковременную эффективность и переносимость.
Карбамазепин**, фенитоин**, фенобарбитал** многие десятилетия использовались в качестве основных ПЭП у больных с фокальной эпилепсией в виде монотерапии. Не смотря на то, что отсутствуют рандомизированные клинические исследования, отражающие их эффективность в качестве дополнительной терапии при фокальных фармакорезистентных эпилепсиях, по мнению авторов, их назначение является обоснованным.
Рандомизированных клинических исследований, отражающих клиническую эффективность и переносимость клоназепама в качестве дополнительного ПЭП у взрослых пациентов с фокальными формами эпилепсии, нет [296].
Применение трех и более ПЭП может быть осуществлено лишь в единичных случаях при резистентных формах эпилепсии и должно быть строго аргументировано.
3.1.1.2.2. Лечение фармакорезистентной генерализованной эпилепсии (генерализованных приступов)
· Рекомендуется назначать в качестве дополнительного средства у взрослых пациентов с фармакорезистентной генерализованной эпилепсией вальпроевую кислоту** ламотриджин, леветирацетам**, перампанел**, топирамат** с целью прекращения приступов [297, 324].
Уровень убедительности рекомендаций В, (уровень достоверности доказательств – 1).
Комментарии: При фармакорезистентности у пациентов с идиопатической генерализованной эпилепсией возникают определённые проблемы с выбором ПЭП. Существует мало ПЭП, лицензированных для лечения данных форм. Вальпроевая кислота**, показавшая высокую эффективность при всех генерализованных приступах, остается препаратом первой очереди выбора для лиц мужского пола.
В основе тезиса-рекомендации лежит первый системный обзор и мета-анализ РКИ [297], в которых сравнивались ПЭП с плацебо и друг с другом при фармакорезистентной генерализованной эпилепсии. Оценивали: снижение приступов на 50% и более, достижение ремиссии приступов и побочные эффекты используемых ПЭП. Этот мета-анализ иллюстрирует, что все проанализированные ПЭП доказывают свою эффективность при фармакорезистентной генерализованной эпилепсии. Данные о влиянии перампанела на миоклонии и абсансы отсутствуют.
3.1.1.3. Лечение при неуточненной форме эпилепсии (недифференцированных приступах)
При невозможности врача определиться фокальная или генерализованная форма эпилепсии у пациента устанавливается диагноз - неуточненная эпилепсия. В этом случае рекомендовано назначение ПЭП с широким спектром действия.
· Рекомендуется назначать взрослым пациентам с неуточненной формой эпилепсии (недифференцированных приступах) вальпроевую кислоту** с целью прекращения приступов [284, 298].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 2).
Комментарии: вальпроевая кислота** предлагается в качестве первой линии для пациента, чьи приступы трудно классифицировать как фокальные или генерализованные в начале лечения, с учетом предполагаемого широкого спектра действия вальпроевой кислоты**. Вальпроевая кислота** лучше переносится, чем топирамат** и более эффективна, чем ламотриджин. Вальпроевая кислота** должна оставаться препаратом первого выбора для многих пациентов с неуточненной эпилепсией. Однако, из-за потенциальных побочных эффектов, высоких тератогенных рисков от назначения вальпроевой кислоты** у девочек и женщин детородного возраста следует отказаться [284].
В рандомизированном клиническом исследовании SANADII при сравнении вальпроевой кислоты** и леветирацетама** у больных с впервые диагностированной генерализованной и неклассифицированной эпилепсией было показано, что леветирацетам** уступает по клинической эффективности вальпроевой кислоте** [298].
l Рекомендуется назначать взрослым пациентам с неуточненной формой эпилепсии (недифференцированных приступах) ламотриджин, леветирацетам**, топирамат** с целью прекращения приступов [299].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
Лечение является более сложным, чем у взрослых пациентов, из-за возрастных ограничений к применению противоэпилептических препаратов, а также в связи с тем фактом, что выбор препарата часто определяется наличием специфического эпилептического синдрома детства.
l Рекомендуется применение фенобарбитала** в качестве монотерапии и в качестве дополнительного препарата ребенку с эпилепсией с периода новорожденности и старше c целью лечения эпилепсии [300].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 1).
Комментарии: Фенобарбитал** является препаратом первой очереди выбора в лечении неонатальных судорог и неонатальных эпилепсий. Его применение у новорожденных в РФ существенно затруднено в связи с отсутствием регистрации формы для внутривенного введения, которая необходима для достижения дозы насыщения. Может применяться и в более старшем возрасте (при всех типах приступов, кроме абсансов), но только при отсутствии эффекта от других противоэпилептических препаратов. Последнее ограничение связано с тем, что препарат может вызывать необратимое ухудшение когнитивных функций у детей, длительно его принимающих [301]. В случае применения препарата в неонатальном периоде и в первые месяцы жизни (при необходимости продолжения лечения эпилепсии) в дальнейшем рекомендуется перевод ребенка на другие противоэпилептические препараты, не вызывающие серьезных когнитивных побочных эффектов. Применение в неонатальном периоде и в первые месяцы жизни затруднено из-за отсутствия специальных детских лекарственных форм, позволяющих точно подобрать дозу препарата.
В комбинации с остальными ПЭП необходимо учитывать возможность серьезного снижения их концентрации за счет активации фенобарбиталом** ферментов печени.
l Рекомендуется применение фенитоина** ребенку с эпилепсией с началом в периоде новорожденности и старше c целью лечения эпилепсии [300].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 1).
Комментарии: Наряду с фенобарбиталом** фенитоин** является препаратом первой очереди выбора в лечении неонатальных судорог и эпилепсий с неонатальным началом. Применение фенитоина** у новорожденных в РФ существенно затруднено в связи с отсутствием регистрации формы для внутривенного введения, которая необходима для достижения дозы насыщения.
В Российской Федерации у детей вне неонатального возраста препарат применяется редко, что, возможно, связано с его потенциальным неблагоприятным влиянием на когнитивные функции [301]. Тем не менее, в других странах (например, США) он применяется довольно широко.
Другие противоэпилептические препараты при отсутствии эффекта от выше указанных фенобарбитала** и фенитоина** также используются в лечении эпилепсии у новорожденных, но они назначаются вне возрастных показаний («out of label»), и, соответственно требуется решение врачебной комиссии для их назначения.
· Рекомендуется применение клоназепама** как дополнительного препарата ребенку с типичными и атипичными абсансами, атоническими и миоклоническими приступами, инфантильными спазмами, фокальными приступами, первично- и вторично-генерализованными типами приступов с периода новорожденности и старше с целью лечения эпилепсии [302].
Уровень убедительности рекомендаций B (уровень достоверности доказательств – 2).
Комментарии: В силу проблем с переносимостью препарат применяется не в монотерапии эпилепсии, а в основном у пациентов с фармакорезистентной эпилепсией в качестве дополнительного [286]. При продолжительном применении клоназепама** в педиатрической практике следует оценивать риск и пользу из-за возможности побочного неблагоприятного действия на физическое и психическое развитие ребенка, в т.ч. отсроченного (может не проявляться в течение нескольких лет).
l Рекомендуется применение окскарбазепина** в качестве первой монотерапии , а также в качестве дополнительного препарата пациенту с фокальными эпилептическими приступами в возрасте 1 месяца и старше с целью лечения эпилепсии [303, 304, 550].
Уровень убедительности рекомендаций для монотерапии и дополнительной терапии для детей в возрасте от 1 месяца до 4 лет В (уровень достоверности доказательств – 3).
Уровень убедительности рекомендаций Для монотерапии и дополнительной терапии для детей в возрасте от 4 лет и старше А (уровень достоверности доказательств – 2).
Комментарии: У ребенка дошкольного возраста целесообразно применении специальной лекарственной формы – суспензии, не только из-за удобства применения, но и из-за возможности подбора более точной дозировки препарата.
l Рекомендуется применение карбамазепина** в качестве первой монотерапии [275], а также в качестве дополнительного препарата пациенту с фокальными приступами с вторичной генерализацией или без нее у детей с целью лечения эпилепсии [305].
Уровень убедительности рекомендаций для монотерапии A (уровень достоверности доказательств – 1).
Для комбинированной терапии: Уровень убедительности рекомендаций фокальных приступов В (уровень достоверности доказательств – 2).
Комментарии: Целесообразен прием лекарственных форм с пролонгированным высвобождением активного вещества, позволяющим осуществлять прием 2 раза в день. В комбинированной терапии следует избегать комбинации с ламотриджином (из-за противосудорожного антагонизма и увеличения числа нейротоксических эффектов). В комбинация с остальными препаратами необходимо учитывать возможность серьезного снижения их концентрации за счет активации ферментов печени карбамазепином**.
Изолированные генерализованные тонико-клонические приступы у детей встречаются редко, если встречаются, то в комбинации с другими типами генерализованных приступов (типичными и атипичными абсансами, миоклониями). Известно, что карбамазепин** может аггравировать течение абсансов, поэтому у детей в лечении генерализованных тонико-клонических приступов карбамазепин** лучше не использовать.
· Рекомендуется применение вальпроевой кислоты** у детей с фокальными, генерализованными и неклассифицированными типами эпилептических приступов в качестве монотерапии и дополнительной терапии в возрасте с 6 месяцев и старше с целью лечения эпилепсии [306].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
Комментарии: У детей, которые не могут глотать таблетки, целесообразно применение специальных лекарственных форм вальпроевой кислоты** (капель, сиропа, микрогранул). У пациентов любого возраста целесообразен прием вальпроевой кислоты** в виде лекарственных форм пролонгированного действия, что улучшает переносимость и дает дополнительный противосудорожный эффект. Следует с осторожностью назначать препарат в группе детей, особенно раннего детского возраста, если у них подозревается наличие митохондриального заболевания. До наступления половой зрелости необходимо постоянно рассматривать возможность переключения пациенток с вальпроевой кислоты** на альтернативные методы лечения (подробнее о назначении препарата у лиц женского пола – см. инструкцию к препарату).
l Рекомендуется применение леветирацетама** у детей с фокальными эпилептическими приступами (с вторичной генерализацией или без нее) с возраста 1 месяц
· Рекомендуется специалистам по реабилитации у больных эпилепсией старше 18 лет учитывать сниженную кардиореспираторную толерантность, с целью оценки модифицируемых факторов развития физических и психических хронических состояний и преждевременной смертности [437, 438].
Уровень убедительности рекомендаций С (уровень достоверности доказательств - 2).
· Рекомендуется проведение акупунктуры в сочетании с основной терапией для пациентов с эпилепсией для снижения частоты приступов [439].
Уровень убедительности рекомендаций А (уровень достоверности доказательств - 1).
· Рекомендуется проведение «школы эпилепсии» с обзором отдельных тем, связанных с эпилепсией, таких как типы приступы, управление стрессом, меры профилактики травм, эффективность лечения и побочные эффекты ПЭП членами междисциплинарной команды для пациентов с эпилепсией и их родственников для улучшения качества жизни пациентов с эпилепсией [440].
Уровень убедительности рекомендаций С (Уровень достоверности доказательств - 5).
· Рекомендуется организация наблюдения, включающего структурированное интерактивное обучающее приложение, организованное обученной медсестрой по эпилепсии, для пациентов с эпилепсией, для улучшения качества жизни, связанного со здоровьем, уменьшения дистресса, связанного с эпилепсией, беспокойства в связи с физическими и психическими эффектами ПЭП, а также в связи с физической активностью и работой [442, 443].
Уровень убедительности рекомендаций С (Уровень достоверности доказательств - 2).
· Рекомендуется использование навык-ориентированных психологических вмешательств (когнитивно-поведенческую терапию (КПТ), включая методы КПТ, такие как дыхательные приемы, рассуждение или визуализация; консультирование; и упражнения на осознанность) у взрослых и подростков с эпилепсией для улучшения качества жизни [443].
Уровень убедительности рекомендаций А. (Уровень достоверности доказательств - 2).
· Рекомендуется специалистам по реабилитации включить поведенческие методы в виде программ самоуправления в дополнении к стандартной терапии в комплексную помощь пациентам с эпилепсией с целью улучшении качества жизни, когнитивных функций и приверженности к лечению [444].
Уровень убедительности рекомендаций С (уровень достоверности доказательств - 5).
Комментарий: Когнитивно-повенденческая терапия представляется важным дополнением к стандартной противоэпилептической терапии при эпилепсии, особенно у пациентов с тревожно-депрессивными расстройствами [445]. Основной задачей когнитивно-поведенческой терапии при эпилепсии является коррекция иррациональных установок мышления пациентов, улучшение навыков совладания, самоэффективности и самоконтроля [446]. Программы когнитивно-поведенческой терапии включают обучение пациентов регулированию настроения, разрешению конфликтующих мыслей и эмоций («когнитивный диссонанс»), управлению стрессом и изменению образа жизни, чтобы минимизировать триггеры приступов [444]. Пациенты учатся определять неадаптивные модели мышления и заменять их более здоровыми когнитивными и поведенческими реакциями [447]. Обучение пациентов «эффективному самоуправлению» сводится к трем направлениям: осторожность изо дня в день, отрегулированный стиль жизни, информированное сознание [448].
· Рекомендуется рациональная психотерапия [449, 450].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4).
Комментарий: Психотерапия при эпилепсии используется для влияния не только на патологические проявления, но и на личность пациента с целью приспособления к повседневной жизни в условиях изменившегося состояния здоровья. Психотерапевтические методы могут способствовать пониманию и смягчению аффективной напряженности, общему успокоению, формированию позитивного настроя у пациентов с эпилепсией. Психотерапия при эпилепсии позволяет более дифференцированно, гибко и «экономно» разрешать вопросы долгосрочного планирования медикаментозной терапии и реабилитации. Психотерапия при эпилепсии применяется в составе комплексного лечения при обязательном условии регулярной медикаментозной терапии [449]. Психотерапевтическая коррекция должна применяться адекватно клинической картине заболевания и с обязательным учетом личности пациента. Главными задачами рациональной психотерапии в виде беседы являются установление доверительного контакта с пациентом и разъяснительное подкрепление всех видов лечебных процедур и лекарственных назначений; изменение представления пациента о своей болезни; коррекция социальной установки, отношения к труду, окружающим. Во время психотерапевтической беседы необходимо обсуждать вопросы: как пациенты должны вести себя в обществе; подчеркивать необходимость участия их в трудовой деятельности; в виде советов рекомендовать, каким образом они могут учиться или работать [450].
4.2. Санаторно-курортное лечение, методы медицинской реабилитации, основанные на использовании природных лечебных факторов
В соответствии с приказом Министерства здравоохранения Российской Федерации от 28 сентября 2020 г. N 1029н «Об утверждении перечней медицинских показаний и противопоказаний для санаторно-курортного лечения» противопоказаниями к санаторно-курортному лечению являются [451]:
- Эпилепсия с текущими приступами, в том числе резистентная к проводимому лечению.
- Эпилепсия с ремиссией менее 6 месяцев (для санаторно-курортных организаций не психоневрологического профиля).
По разным оценкам, до 25% случаев развития эпилепсии можно предотвратить путем первичной профилактики заболеваний и состояний, которые могут быть факторами риска эпилепсии. Предупреждение травм головы является самым эффективным способом предотвращения посттравматической эпилепсии. Надлежащая перинатальная помощь способствует уменьшению числа новых случаев эпилепсии, обусловленных перинатальной патологией ЦНС. Необходимы профилактиктические мероприятия, направленные на снижение заболеваемости и раннюю диагностику инфекционных и паразитарных заболеваний, являющихся факторами риска инфекционных эпилепсий.
· Рекомендуется генетическое консультирование и молекулярно-генетическое исследование мутации методом Сэнгера родителям пациента при выявлении у ребенка вероятно патогенной мутации или мутации с неясным клиническим значением в гене, ассоциированным с моногенным заболеванием, с целью прогноза деторождения [256].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5).
· Рекомендуется проведение профилактических мероприятий, направленных на снижение риска сердечно-сосудистых и цереброваскулярных заболеваний, а также диспансерное наблюдение за пациентами, перенесшими инсульт (контроль артериального давления, холестерина, глюкозы крови, снижение веса, а также отказ от курения и чрезмерного употребления алкоголя), для снижения риска постинсультной эпилепсии [452, 453].
Уровень убедительности рекомендаций С (уровень достоверности доказательств - 5).
· Рекомендуется диспансерное наблюдение пациентов с эпилепсией врачом-неврологом поликлиники или специализированного эпилептологического центра, проведение длительной противоэпилептической терапии с целью достижения ремиссии заболевания, сохранения оптимального качества жизни [6].
Уровень убедительности рекомендаций С (уровень достоверности доказательств — 5).
Медицинская помощь по профилю «эпилептология» осуществляется в виде:
- первичной медико-санитарной помощи;
- скорой медицинской помощи;
- специализированной медицинской помощи,
- высокотехнологичной медицинской помощи.
Медицинская помощь оказывается в следующих условиях:
- вне медицинской организации (по месту вызова бригады скорой медицинской помощи);
- амбулаторно (в условиях, не предусматривающих круглосуточное медицинское наблюдение и лечение);
- стационарно (в условиях, обеспечивающих круглосуточное медицинское наблюдение и лечение).
Первичная медико-санитарная помощь включает: первичную врачебную медико-санитарную помощь, первичную специализированную медико-санитарную помощь.
Первичная врачебная медико-санитарная помощь при эпилепсии в соответствии с Приказом Минздрава России от 24.12.2012 № 1404н [454] и Приказом Минздрава России от 24.12.2012 № 1439н [455] оказывается врачом-кардиологом, врачом-неврологом, врачом-психиатром, врачом-терапевтом, врачом-акушером-гинекологом, врачом-нейрохирургом, врачом-офтальмологом, врачом-эндокринологом, врачом-генетиком, врачом общей практики (семейным врачом), врачом-терапевтом участковым, врачом-эндокринологом в медицинских организациях.
Первичная специализированная медико-санитарная помощь при эпилепсии в соответствии с Приказом Минздрава России от 24.12.2012 № 1541н [456] оказывается врачом-акушером-гинекологом, врачом-генетиком, врачом-кардиологом, врачом-неврологом, врачом-нейрохирургом, врачом-офтальмологом, врачом-психиатром, врачом-терапевтом, врачом-эндокринологом в медицинских организациях, оказывающих специализированную, в том числе высокотехнологичную, медицинскую помощь.
Скорая медицинская помощь оказывается выездными бригадами скорой медицинской помощи в соответствии Приказом Минздрава России от 05.07.2016 N 468н [457]. Обязательной госпитализации подлежат пациенты с эпилептическим статусом, в состоянии комы и в состоянии с нарушением сознания в медицинские организации, оказывающие круглосуточную помощь и имеющие в своем составе отделения неотложной медицинской помощи.
Высокотехнологичная медицинская помощь осуществляется в соответствии с Приказом Министерства здравоохранения РФ от 10 декабря 2013 г. № 916н "О перечне видов высокотехнологичной медицинской помощи"[458].
При обращении пациента с эпилепсией к врачу общей практики, врачу-терапевту, кардиологу он направляется на консультацию к врачу-неврологу для определения плана обследования и назначения лечения. Большинство пациентов с эпилепсией могут проходить обследование и лечение у врача-невролога. Пациенты с эпилепсией состоят под наблюдением у врачей-неврологов.
В межрайонные (районные) специализированные кабинеты оказания помощи больным с эпилепсией и другими пароксизмальными состояниями и/или городские эпилептологические центры и/или областные эпилептологические центры (кабинеты) направляются больные для уточнения диагноза, назначения лечения и его коррекции.
Отбор больных для проведения консультаций в межрайонные (районные) специализированные кабинеты оказания помощи больным с эпилепсией и другими пароксизмальными состояниями, городские эпилептологические центры, областные эпилептологические центры (кабинеты) осуществляет врач-невролог или заведующий неврологического отделения медицинской организации.
Направлению в межрайонные (районные) специализированные кабинеты оказания помощи больным с эпилепсией и другими пароксизмальными состояниями и/или городские эпилептологические центры, областные эпилептологические центры (кабинеты) подлежат пациенты: с впервые выявленной эпилепсией; с ранее установленным диагнозом эпилепсии для коррекции лечения; при сохранении приступов на протяжении более 3 месяцев на фоне назначенной терапии [459]; с установленной резистентностью к проводимой терапии; с целью дифференциального диагноза эпилепсии с другими пароксизмальными состояниями; лица призывного возраста, имеющие в анамнезе пароксизмальные состояния; женщины с пароксизмальными состояниями, планирующие беременность.
При сохранении эпилептических приступов, развитии побочных эффектов, фармакорезистентных формах заболевания пациент может быть направлен в неврологический стационар; по показаниям в нейрохирургический стационар.
В неврологическом стационаре проводится поиск возможных причин «псевдорезистентности», проводится дифференциальный диагноз с псевдоэпилептическими приступами, пароксизмальными нарушениями сознания другой этиологии, с привлечением врачей других специальностей, уточняются типы эпилептических приступов, форма эпилепсии, осуществляется коррекция терапии, решается вопрос о возможности проведения «кетогенной диеты»,необходимости и возможности прехирургической подготовки и хирургического лечения пациентов с фармакорезистентным течением заболевания.
В стационаре должна быть возможность проведения нейрофизиологических методов обследования (ЭЭГ, видео-ЭЭГ-мониторинга), специализированного нейропсихологического тестирования, методов структурной нейровизуализации (МРТ-головного мозга, КТ головы), консультаций врачей специалистов (врача-психиатра, врача-кардиолога, врача-нейрохирурга и др). Только в условиях стационара возможно проведения видео-ЭЭГ-мониторинга со снижением доз либо отменой ПЭП с целью регистрации приступа (пароксизмального события) в случае проведения прехирургической подготовки пациентов с фармакорезистентными формами эпилепсии. При снижении доз ПЭП должна быть возможность оказания медицинской помощи в случае развития эпилептического статуса.
В нейрохирургический стационар направляются пациенты с фармакорезистентными формами эпилепсии для проведения прехирургической подготовки, в том числе с применением инвазивных электродных систем, и решения вопроса о нейрохирургическом лечении (резективных оперативных вмешательств, установке стимулятора блуждающего нерва и др.), проведении высокотехнологичной медицинской помощи.
Пациент с впервые выявленным эпилептическим приступом и пароксизмальным нарушением сознания подлежит обследованию и лечению в условиях стационара. С учётом конкретных клинико-анамнестических и данных рутинных лабораторно-инструментальных исследований врачу приемного отделения следует определить дальнейшую маршрутизацию пациента. При выявлении признаков эпилептического приступа, другой органической патологии центральной нервной системы показано продолжение обследования и лечения в условиях отделения неврологии. При выявлении признаков эпилептического статуса следует продолжить лечение в условиях отделения реанимации и интенсивной терапии.
Предположение психогенной природы пароксизмального события требует осмотра психиатра.
При веских доводах в пользу синкопального состояния пациент осматривается врачом-терапевтом (кардиологом) и направляется на лечение в терапевтическое (кардиологическое) отделение.
При наличии у пациента угрожающих нарушений сердечного ритма, гемодинамической нестабильности следует продолжить лечение в условиях отделения интенсивной терапии. По показаниям, осуществляется консультация врача-сердечно-сосудистого хирурга. При наличии обтурирующих интракардиальных образований, острой сердечно-сосудистой патологии пациента следует направить в отделение кардиохирургии или рентген-эндоваскулярных методов лечения.
