














Неонатальный холестаз – нарушение экскреторной функции гепатобилиарной системы в периоде новорождённости, приводящее к повышению компонентов желчи в крови и их дефициту в кишечнике [1–3].
Синонимы: гипербилирубинемия новорождённых, обусловленная нарушением экскреции билирубина; прямая гипербилирубинемия новорождённых; синдром сгущения желчи.
Морфофункциональные особенности печени и желчных протоков у новорожденных детей характеризуются высоким уровнем синтеза желчных кислот и незрелостью их печеночно-кишечной циркуляции, предрасполагают к нарушению оттока желчи. У новорожденных детей отмечается несоответствие между повышенной продукцией билирубина, желчных кислот и других компонентов желчи и ограниченной способностью к их выведению из организма. В норме на 4-7 сутки жизни образуется намного больше желчных кислот, чем у взрослых. Желчные кислоты новорожденных называются «атипичными» в связи с наличием на их поверхности дополнительных гидроксильных групп, уменьшающих индекс их токсичности, и вместе с тем значительно снижающих их холекинетическую активность. Относительная незрелость ферментных систем печени, обеспечивающих захват компонентов желчи из крови, их внутриклеточный транспорт и экскрецию во внутрипеченочную желчную систему, повышенная проницаемость межклеточных соединений, низкая экскреторная активность по желчевыводящей системе и повышенная реабсорбция компонентов желчи в кишечнике способствуют медленному выведению желчи и повышенному содержанию желчных кислот и других компонентов желчи в крови. Это проявляется у новорожденных детей относительным увеличением размеров печени (до 2 см ниже реберной дуги), повышением прямой фракции билирубина до 15-20% от уровня общего, более высоким содержанием в крови ферментов – ЩФ, ГГТ и желчных кислот и может рассматриваться в этот период как пограничное состояние - физиологический холестаз новорожденных. Полное становление механизмов образования желчи и ее печеночно-кишечной циркуляции заканчивается после месячного возраста, достигая уровня взрослого к 3-6 месяцам жизни [4–6].
Причиной неонатального холестаза может быть:
– Внепечёночная перинатальная патология.
– Заболевания печени и желчевыводящих протоков.
Основные заболевания, проявляющиеся неонатальным холестазом представлены в таблице 1.
Таблица 1. Заболевания печени и желчных протоков, проявляющиеся синдромом холестаза у новорожденных детей.

В соответствующих действующих клинических рекомендациях подробно описаны такие заболевания как Галактоземия [7], Тирозинемия [8], Болезнь Ниманна-Пика тип С [9], Муковисцидоз [10] и Гипотиреоз [11].
Причинами синдрома холестаза в неонтальном периоде могут быть как транзиторные состояния, связанные с тяжелой перинатальной патологией и инфекции, проявляющиеся гепатитом, так и ряд врожденных заболеваний печени вследствие аномалий развития желчевыводящей системы, нарушения синтеза и/или транспорта компонентов желчи и метаболические заболевания. Некоторые заболевания, наиболее часто встречающиеся и/или имеющие эффективные методы лечения, представлены ниже.
1. Неонатальный холестаз, обусловленный внепечёночной перинатальной патологией – нарушение экскреторной функции гепатобилиарной системы, вызванное совокупностью патологических и ятрогенных факторов перинатального периода, а также морфофункциональной незрелостью печени. В структуре внепечёночных причин формирования неонатального холестаза ведущее место занимают состояния, сопровождающиеся развитием гипоксии или ишемии гепатобилиарной системы, гипоперфузией желудочно-кишечного тракта (ЖКТ), стойкой гипогликемией метаболическим ацидозом и застойной сердечно-сосудистой недостаточностью. Нарушение экскреторной функции гепатобилиарной системы может быть обусловлено повышенным содержанием билирубина при ГБН, вследствие значительного изменения коллоидных свойств желчи, повышением её вязкости, а в ряде случаев вследствие непосредственного токсического действия билирубина на мембраны гепатоцитов и митохондрии клеток. Важное место занимают системные и локализованные бактериальные инфекции, запускающие синтез и экскрецию сложного каскада медиаторов воспаления купферовскими клетками, а также гепатоцитами и эндотелиальными клетками синусоидов, оказывающих непосредственное влияние на образование и экскрецию жёлчи. Лечебные мероприятия, проводимые новорождённым в условиях отделения реанимации и интенсивной терапии новорождённых (ОРИТН), включают потенциально гепатотоксичные лекарственные средства, средства для парентерального питания (ПП). Они также способствуют нарушению функционального состояния гепатобилиарной системы. Развитие холестаза чаще отмечают у недоношенных новорождённых при одновременном действии нескольких патологических и ятрогенных факторов на функцию печени и состояние жёлчных протоков. В основе этих изменений лежат различные деструктивные изменения желчевыводящих протоков, нарушения проницаемости мембран гепатоцитов и межклеточных соединений (в большинстве случаев обратимые). Формирование неонатального холестаза на фоне внепечёночной перинатальной патологии характерно для периода новорождённости, так как именно в этом возрасте существует морфофункциональная незрелость гепатобилиарной системы – результат сочетанного действия патологических и ятрогенных факторов [1,4,12–14].
2. Заболевания печени и желчевыводящих протоков.
В зависимости от уровня поражения гепатобилиарной системы принято выделять болезни, проявляющиеся внепечёночным и внутрипечёночным холестазом.
Причинами внепечёночного холестаза у новорождённых могут быть:
Билиарная атрезия (БА).
Киста общего желчного протока.
«Желчные пробки» или камни желчного протока.
Сдавление общего желчного протока.
2.1 Билиарная атрезия (БА) – прогрессирующая облитерация внепечёночных желчных протоков, начинающаяся в период внутриутробного развития, с постепенным вовлечением в процесс внутрипечёночной желчной системы и формированием билиарного цирроза. В настоящее время этиология не ясна. Обсуждают теорию порока развития, вирусную, генетическую и другие. Отсутствие проходимости жёлчных путей приводит к накоплению компонентов желчи в гепатобилиарной системе и повышенному их поступлению в кровь. Желчь не поступает в кишечник и, как следствие, нарушаются процессы переваривания и всасывания жиров и жирорастворимых витаминов. Определённую роль в патогенезе БА играет иммунная система. При проведении цитохимического исследования биоптата печени выявляют клеточные маркёры воспаления, и, в том числе, СD14-положительные макрофаги, запускающие выработку каскада иммунологических реакций. Экспрессия внутриклеточных адгезивных молекул I типа способствует формированию лейкоцитарных антигенов вокруг жёлчных протоков, что в свою очередь запускает цитотоксическую «лимфоцитарную атаку». Процесс воспаления эпителиальных клеток жёлчных протоков сопровождается активной выработкой ростового фактора, стимулирующего транскрипцию коллагена I типа, лежащего в основе перидуктального фиброза. Выделяют четыре типа заболевания: I тип (3%) – атрезия только общего жёлчного протока; II тип (6%) – киста в воротах печени, соединённая с внутрипечёночными жёлчными протоками; III тип (19%) – атрезия левого и правого печёночных протоков, жёлчный пузырь, пузырный и общий жёлчный протоки проходимы; IV тип (72%) – атрезия всей внепечёночной системы [15,16].
2.2 Киста общего желчного протока – врождённое расширение желчного протока, которое в 2–5% случаев вызывает полное нарушение проходимости желчевыводящих протоков и может быть причиной внепечёночного холестаза. Первичные кисты общего желчного протока обусловлены изначальным истончением или отсутствием мышечной стенки и замещением её соединительной тканью. Вторичные – пороком развития, формирующимся в период обратного развития солидной стадии эмбриогенеза (в период 3–7 нед.). Возникающие при этом перегибы, стенозы или клапаны конечного отдела общего желчного протока приводят к его расширению, истончению стенки. Характерны изолированные расширения только общего желчного протока без вовлечения в процесс пузырного протока и стенки желчного пузыря. Возможно также аномальное соединение кисты с панкреатическим протоком; считается, что заброс панкреатических ферментов в общий желчный проток способствует развитию кисты. При сочетании кисты общего желчного протока с нарушением проходимости желчной системы патогенез не отличается от такового при БА. В остальных случаях застой желчи в расширенном желчном протоке приводит к изменению её коллоидных свойств, повышенной вязкости и неполному оттоку желчи. Длительное сохранение желчи в протоках предрасполагает к инфекционным осложнениям с развитием холангита.
2.3. Неонатальный гепатит (врождённый гепатит, фетальный гепатит) – патология печени инфекционной природы в перинатальном и постнатальном периоде. Вирусы (цитомегалловирус, вирус краснухи, вирус герпеса, Коксаки и другие, вирусы гепатита В, С), бактерии (листерии, Micobacterium tuberculosis и другие), простейшие (токсоплазма), а также Treponema pallidum и др. могут стать причиной гепатита. Гепатит, резвившийся на фоне течения неонатального сепсиса, чаще вызван теми же, что и сепсис, возбудителями. Высокий риск заражения плода и новорождённого гепатитом В существует при следующих условиях: развитие острого гепатита В у матери в III триместре беременности; носительство матерью HBSAg в сочетании с положительным HBEAg и отсутствием анти HBE; выявлением ДНК гепатита В методом ПЦР. высоким титром анти-HBCOR. В основе патогенеза лежит специфическая воспалительная реакция в ответ на инфицирование микроорганизмами, тропными к гепатоцитам. Неонатальный холестаз цитомегаловирусной этиологии подробно представлен в клинических рекомендациях «Врожденная цитомегаловирусная инфекция» [17].
2.4 Синдром Алажилля (синдромальная форма гипоплазии внутрипечёночных желчных протоков, артериопечёночная дисплазия) – синдромальная форма патологии, включающая сочетание не менее трёх из пяти основных признаков: хронический холестаз, в основе которого лежит врождённая гипоплазия внутрипечёночных желчных протоков, сердечно-сосудистые дефекты, аномалии позвоночника, дефекты глаз, характерные черепно-лицевые признаки [18]. Синдром Алажилля имеет аутосомно-доминантный тип наследования. Генный дефект связан с частичной делецией короткого плеча 20 хромосомы [20р11-12] где локализуется Jagged1 (JAG1) ген. Реже выявляется мутации в другом гене, называемом NOTCH2. В основе изменений печени при синдроме Алажилля лежит врождённая гипоплазия внутрипечёночных желчных протоков, степень выраженности которой может широко варьировать и определять как время появления первых клинических симптомов, так и, прогноз заболевания. Гипоплазия внутрипечёночных желчных протоков затрудняет отток желчи, что приводит к накоплению её компонентов в гепатобилиарной системе и повышенному поступлению в кровь. С другой стороны, недостаточное поступление желчи в кишечник приводит к нарушению процессов всасывания жиров и жирорастворимых витаминов.
2.5. Несиндромальная форма гипоплазии внутрипеченочных желчных протоков устанавливается при отсутствии характерных для синдрома Алажилля аномалий и/или пороков развития, патогенных вариантов генах JAG1 и NOTCH2, а также при выявлении гипоплазии при гистологическом исследовании биоптата печени. Этиология данного заболевания не установлена, существует предположение о сочетанном действии целого ряда тератогенных факторов на закладку и формирование внутрипеченочных желчных протоков. Следует также отметить, что данный гистологический признак может быть одним из проявлений других заболеваний, и в том числе инфекционных болезней (краснухи, ЦМВ и сифилиса), метаболических нарушений (дефицита а-1-антитрипсина, муковисцидоза), эндокринных нарушений (гипопитуитаризма) и хромосомных заболеваний (трисомия 18 и 21 хромосом). В связи с этим при выявлении гипоплазии внутрипеченочных желчных протоков следует обратить внимание на характерные для этих поражений клинико-лабораторные признаки и провести дополнительные исследования. Исключение этих заболеваний служит основанием для установления несиндромальной (идиопатической) гипоплазии внутрипеченочных желчных протоков.
2.6. Прогрессирующий семейный внутрипеченочный холестаз (ПСВХ) – это класс хронических холестатических заболеваний печени, обусловленных генетически-детерминированным нарушением структуры канальцевой мембраны гепатоцита, приводящим к нарушению экскреции желчных кислот и/или других компонентов желчи. На сегодняшний момент выделяют до 12 типов ПСВХ (Таблица 2) [19–27]. Тем не менее наиболее изученными являются 5 типов: 1 тип (болезнь Байлера), 2 тип (синдром Байлера), 3 тип (дефицит MDR3-гена), 4 тип (дефицит TJP2 гена), 5 тип (дефицит фарнезоидного рецептора Х).
Таблица 2. Типы ПСВХ
Тип |
Белок (ген) |
Функция белка |
|---|---|---|
ПСВХ-1 |
FIC1 (ATP8B1) |
Поддержание амминофосфолипидов мембран клеток и, в том числе канальцевой мембраны гепатоцитов, ответственных за экскрецию желчных кислот и других компонентов желчи. |
ПСВХ-2 |
BSEP (ABCB11) |
Экскреция желчных кислот из гепатоцитов в желчные протоки |
ПСВХ-3 |
MDR3 (ABCB4) |
Отвечает за транспорт фосфотидилхолина через канальцевую мембрану гепатоцита |
ПСВХ-4 |
TJP2 (TJP2) |
Соединяет трансмембранные белки плотных контактов с актиновым цитоскелетом, необходимые для экскреции желчных кислот |
ПСВХ-5 |
FXR (NR1H4) |
Транспортировка белка BSEP из ЭПР в плазматическую мембрану, ответственного за экскрецию желчных кислот |
ПСВХ-6 |
SLC51A/OSTα(SLC51A) |
Реабсорбции желчных кислот в кишечнике |
ПСВХ-7 |
USP53 (USP53) |
Является белком плотных контактов TJP1 и 2, участвующих в экскреции желчных кислот |
ПСВХ-8 |
KIF12 (KIF12) |
Регулирует полярность гепатоцитов и процессы внутриклеточного транспорта. |
ПСВХ-9 |
ZFYVE19 (ZFYVE19) |
Неизвестно, может регулировать полярность ресничек и холангиоцитов |
ПСВХ-10 |
MYO5B (MYO5B) |
Учувствует в поляризации эпителиальных клеток, а также локализации BSEP на канальцевой мембране |
ПСВХ-11 |
SEMA7A (SEMA7A) |
Неизвестно, может регулировать экспрессию канальцевых переносчиков желчных кислот |
ПСВХ-12 |
VPS33B (VPS33B) |
Регулирует полярность гепатоцитов и функции желчных канальцев. |
Нарушение экскреции желчных кислот и/или других компонентов желчи приводит к их накоплению в гепатоцитах. Достигая определенной концентрации в гепатоцитах, компоненты желчи выходят в кровь, оказывая при этом токсическое действие. Отсутствие или дефицит ЖК в кишечнике приводит к нарушению процессов всасывания жиров и жирорастворимых витаминов.
2.7 Нарушение синтеза желчных кислот вследствие ферментопатии – группа редких наследственных заболеваний, в основе которых лежит дефицит фермента, принимающего участие в синтезе первичных желчных кислот. В зависимости от дефицита той или иной ферментной системы выделяют 9 заболеваний, для 2-х из которых описана манифестация в неонатальном периоде (таблица 3).
Таблица 3 Врожденные нарушения синтеза желчных кислот, проявляющиеся неонатальным холестазом

Дефицит фермента, принимающего участие в синтезе ЖК, приводит к накоплению промежуточных продуктов их синтеза. Эти соединения не поступают в кишечник и накапливаются в гепатоцитах. Промежуточные продукты синтеза ЖК, достигая определенной концентрации в гепатоцитах, выходят в кровь, оказывая при этом токсическое действие, прежде всего, на гепатоциты. Отсутствие ЖК в кишечнике приводит к нарушению процессов всасывания и, в том числе всасывания жирорастворимых витаминов.
Распространенность синдрома холестаза зависит от этиологии. Эпидемиология некоторых заболеваний указана в Таблице 4 [6]. Суммарная частота встречаемости составляет 1 на 2500 новорожденных [21,28].
Таблица 4. Распространенность некоторых врожденных заболеваний печени и метаболических заболеваний, сопровождающихся неонатальным холестазом.
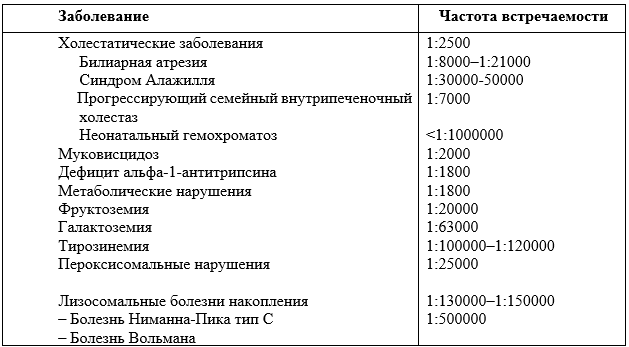

P58.2 Неонатальная желтуха, обусловленная инфекцией
P58.4 Неонатальная желтуха, обусловленная лекарственными средствами или токсинами, перешедшими из организма матери или введенными новорожденному
P59.1 Синдром сгущения желчи
P59.2 Неонатальная желтуха вследствие других и неуточненных повреждений клеток печени
Впоследствии при уточнении причины неонатального холестаза кодирование заболевания трансформируется в соответствующий код выявленной нозологии.
На сегодняшний момент классификации синдрома холестаза у новорожденных отсутствует.
Клинические проявления транзиторного синдрома холестаза включают желтуху с зеленоватым оттенком, увеличение размеров печени, насыщенно жёлтый цвет мочи, эпизоды ахолии стула, развитие которых отмечается на фоне перинатальной патологии. По мере улучшения общего состояния ребенка и разрешения основного заболевания, в большинстве случаев происходит постепенное уменьшение холестаза. Однако остаточные его явления могут сохраняться в течение длительного времени, до 6-8 месяцев жизни.
Неонатальный гепатит в большинстве случаев является одним из проявлений генерализованной инфекции, хотя более чем в 50% случаев при внутриутробной инфекции изменения печени носят реактивный характер. Клинические проявления при гепатите включают нарушение общего состояния, признаки инфекционного процесса, желтуху, увеличение размеров печени и селезёнки, геморрагический синдром и другие признаки TORCH синдрома (Toxoplasmosis (токсоплазмоз), Other infections (другие инфекции, включая сифилис, гепатит, ВИЧ.), Rubella (краснуха), Cytomegalovirus (цитомегаловирус), и Herpes simplex virus (вирус простого герпеса)). Большинство новорождённых, инфицированных вирусом гепатита В или С, формируют бессимптомное носительство в течение всей жизни или первично хронический гепатит.
Пациенты с билиарной атрезией в большинстве случаев рождаются доношенными с антропометрическими показателями, соответствующими физиологической норме. Желтуха появляется на 2-3-и сутки жизни, т.е. в обычные для физиологической желтухи сроки. Примерно у 2/3 больных отмечают наличие «светлого промежутка» уменьшение интенсивности желтухи к концу 1-2-й неделе жизни с последующим постепенным её нарастанием и появлением зеленоватого оттенка кожи к концу 1-го месяца. Ахолия стула – наиболее ранний и постоянный клинический при болезни. Её появлению часто предшествует отхождение мекония. При оценке цвета стула следует также помнить, что при использовании некоторых лечебных смесей с добавлением среднецепочечных триглицеридов, эквивалентом обесцвеченного стула могут быть разные оттенки серого цвета. Характерно для БА отсутствие гепатомегалии при рождении с последующим увеличением размеров печени и изменением её консистенции от эластичной до плотной в течение первых двух месяцев жизни. К возрасту 1 месяца возможно развитие геморрагического синдрома (кровоточивость слизистых ЖКТ, пупочной ранки, внутричерепное кровоизлияние). К возрасту 1-2 месяцев жизни, как правило, формируется дефицит веса, степень выраженности которого зависит от вида вскармливания ребёнка. Наиболее выраженный дефицит отмечают при грудном вскармливании или использовании искусственных смесей, предназначенных для питания здоровых новорождённых. При использовании лечебного питания, высококалорийного, с частичным расщеплением различных составляющих, дефицит веса может отсутствовать или быть минимально выраженным. Без хирургического лечения уже к 5-6 месяцам жизни появляются признаки портальной гипертензии, которая в дальнейшем прогрессивно нарастает и свидетельствует о формировании билиарного цирроза [15,29].
Клинические проявления кисты общего желчного протока можно наблюдать при нарушении проходимости желчных путей или развитии инфекционных осложнений (холангита). В первом случае клинико-лабораторные проявления не отличаются от таковых при БА. При развитии холангита характерно сочетание признаков внепечёночного холестаза с воспалительными изменениями. При этом в клинической картине отмечают повышение температуры тела, ухудшение общего состояния и другие симптомы [30].
При синдроме Алажилля холестаз появляется в период новорождённости, реже в течение первых месяцев жизни. Отмечается желтуха с зеленоватым оттенком кожи, увеличение размеров печени, непостоянная ахолия стула, тёмный цвет мочи. В дальнейшем, после 4-6 месяцев жизни, наблюдают уменьшение или исчезновение желтухи, нормализацию цвета стула и мочи. Однако появляется кожный зуд, который в дальнейшем усиливается и, становится ведущим клиническим симптомов заболевания, тогда как другие проявления имеют перемежающийся характер [31–34].
Клиническими проявлениями прогрессирующего семейного внутрипеченочного холестаза являются желтуха, непостоянная ахолия стула, характерным является раннее, в течение первых месяцев жизни, появление кожного зуда. Большинство из типов ПСВХ характеризуются низкой активностью ГГТ [26,27,35]. Основные характеристики различных типов ПСВХ представлены в таблице 5.
Таблица 5. Основные характеристики прогрессирующего семейного внутрипеченочного холестаза
Тип ПСВХ |
1 тип |
2 тип |
3 тип |
4 тип |
5 тип |
|---|---|---|---|---|---|
Манифестация |
неонатальный период/первые 3 мес. |
неонатальный период/первые 3 мес. |
неонатальный период/ранний возраст |
неонатальный период/первые 3 мес. |
неонатальный период/первые 3 мес. |
Зуд |
сильный |
сильный |
умеренный |
сильный/ умеренный |
сильный/ умеренный |
Уровень ГГТ |
нормальный |
нормальный |
повышен |
нормальный/ немного повышенный |
нормальный |
Уровень холестерина |
нормальный |
нормальный |
повышенный/нормальный |
нормальный |
нормальный |
Пролиферация ЖП |
нет |
нет |
есть |
нет |
есть |
Уровень ЖК в крови |
очень высокий |
очень высокий |
высокий |
высокий |
высокий |
Внепеченочные проявления |
диарея, панкреатит нейросенсорная глухота, низкорослость |
нет |
нет |
неврологические и респираторные нарушения |
диарея |
Риск развития онкологических заболеваний гепатобилиарной системы |
нет |
гепатоцеллюлярная карцинома/ холангиокарцинома |
нет |
гепатоцеллюлярная карцинома |
нет |
При нарушении синтеза желчных кислот появление первых признаков болезни отмечается в периоде новорожденности, реже в течение первых месяцев жизни. Отмечается желтуха, гепатомегалия, темный цвет мочи, непостоянная ахолия стула. Характерна диссоциация между низким уровнем ГГТ и холестерина сыворотки крови и повышением других маркеров холестаза, значительное повышение АЛТ и АСТ. Характерно нарушение белок-синтетической функции печени (снижение альбумина, ХЭ, фибриногена, ПТИ, повышение МНО и других показателей). При морфологическом исследовании биоптата печени отмечаются признаки преимущественно внутрипеченочного холестаза, гигантоклеточная трансформация гепатоцитов и дезорганизация печеночных долек. Характерно также отставание детей в физическом развитии и состояния, связанные с дефицитом жирорастворимых витаминов. Возможно развитие геморрагического синдрома.
Критерии установления диагноза/состояния:
Синдром холестаза вне зависимости от этиологии может быть установлен на основании повышения прямого билирубина более чем на 20% от уровня общего или более 17 мкмоль/л у детей первых 7 суток жизни в сочетании с повышением активности щелочной фосфатазы (ЩФ), гамма-глутамилтрансферазы (ГГТ), холестерина и желчных кислот.
Диагноз «неонатальный холестаз», являющийся осложнением тяжелой внепеченочной патологии, устанавливают при выявлении предрасполагающих к его развитию факторов и исключения болезней печени и желчных протоков.
Рекомендуется провести сбор анамнеза и жалоб при заболеваниях печени и желчевыводящих путей всем пациентам с целью выявления характерных для синдрома холестаза признаков [36].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: у родителей могут быть жалобы на желтуху, обесцвеченный стул, малую прибавку в весе и росте, срыгивания у ребенка.
Рекомендуется провести анализ наследственного анамнеза всем пациентам с целью выявления сходных случаев заболевания у родственников [37].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: при сборе анамнеза необходимо обратить внимание на наличие отягощенного семейного анамнеза (родственный брак, сходные случаи заболевания у родных братьев и сестер); наличие родственников с патологией печени, почечной недостаточностью, а также случаи гибели от печеночной недостаточности в семье.
Рекомендуется провести сбор анамнеза и жалоб терапевтический всем пациентам с целью выявления особенностей течения патологического процесса [36].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: у пациентов с синдромом холестаза в дебюте следует отметить сроки появления желтухи, обесцвеченного стула, гепатомегалии и спленомегалии. При сборе анамнеза выявляют патологические состояния перинатального периода и терапевтические воздействия, способствующие нарушению экскреторной функции печени.
Рекомендуется провести визуальный осмотр терапевтический всем пациентам с целью выявления признаков синдрома холестаза [37].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: при осмотре пациентов оценивают цвет кожного покрова и склер, размеры печени и селезёнки, цвет стула и мочи. Необходимо измерение печени по трем линиям – правая передне-подмышечная, правая среднеключичная и срединная линии. Селезенка пальпируется по левой передне-подмышечной линии. В случае кисты общего желчного протока при пальпации живота в правом подреберье можно определить объёмное образование.
Рекомендуется проведение общего (клинического) анализа крови развернутого всем пациентам с целью выявления патологических изменений [36,37].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: у новорожденных с тяжелой перинатальной патологией, инфекционным поражением печени, а также при развитии гиперспленизма отмечаются анемия, тромбоцитопения и др.
Рекомендуется всем пациентам проведение анализа крови биохимического общетерапевтического (исследование уровня общего белка в крови, исследование уровня альбумина в крови, исследование уровня общего билирубина в крови, исследование уровня билирубина свободного (неконъюгированного) в крови, исследование уровня билирубина связанного (конъюгированного) в крови, исследование уровня креатинина в крови, исследование уровня мочевины в крови, исследование уровня глюкозы в крови, исследование уровня триглицеридов в крови, исследование уровня холестерина в крови, исследование уровня калия в крови, исследование уровня натрия в крови, исследование уровня общего кальция в крови, исследование уровня неорганического фосфора в крови; определение активности лактатдегидрогеназы в крови, определение активности креатинкиназы в крови, определение активности аспартатаминотрансферазы в крови, определение активности аланинаминотрансферазы в крови, определение активности гамма-глютамилтрансферазы в крови, определение активности щелочной фосфатазы в крови, исследование уровня железа сыворотки крови) [13,36–38] [96-97].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: для синдрома холестаза патогномонично повышение маркёров холестаза: уровня прямого билирубина, активности ЩФ, ГГТ, холестерина и желчных кислот. Важно отметить, что показанием для обследования новорожденного для исключения билиарной атрезии и других холестатических заболеваний печени является повышение уровня прямого билирубина выше 17 мкмоль/л у новорожденных до двух недель жизни, вне зависимости от уровня общего билирубина, а у пациентов старше двух недель жизни уровень прямого билирубина > 17 мкмоль/л при уровне общего билирубина менее 85 мкмоль/л или более 20% от уровня общего билирубина при уровне общего билирубина более 85 мкмоль/л. При большинстве типов ПСВХ, а также при врожденных нарушениях синтеза желчных кислот характерна диссоциация между низким уровнем ГГТ и повышением других маркеров холестаза (прямого билирубина, ЩФ).
При транзиторном холестазе у новорожденных часто отмечают отсроченное (по отношению к холестазу) повышение ферментов цитолиза менее чем в 6-8 раз. Отношение АЛТ/АСТ менее 1. Показатели, отражающие синтетическую функцию печени (альбумин, холинэстераза), как правило, не изменяются.
При БА степень повышения маркеров холестаза в динамике нарастает от минимального повышения в течение первых 2-3 недель жизни до значительного повышения к 2-3 месяцам; активность ферментов цитолиза (АЛТ, ACT) при БА повышается умеренно, и, как правило, отсрочено. В большинстве случаев в течение первых 2-3 недель после рождения эти показатели остаются в пределах нормы и затем постепенно повышаются; уровень альбумина, холинэстеразы, отражающие синтетическую функцию печени, на ранних сроках болезни (в течение первых 3-4 мес. жизни) не изменяются.
Для неонатального гепатита инфекционной природы, помимо повышения маркеров холестаза, характерно повышение АЛТ, АСТ более чем в 8–10 раз, соотношение АЛТ/АСТ больше 1; а также нарушение синтетической функции печени: снижение альбумина, холинэстеразы, фибриногена, ПТИ и др. Исследование активности креатинкиназы проводится с целью дифференциального диагноза с заболеваниями, протекающими с синдромом цитолиза.
Исследование мочевины и креатинина проводится с целью исключения поражения почек, которое может сопутствовать наследственным заболеваниям печени, таким, как синдром Алажилля, когда аномалии развития почек нередко приводят к развитию почечной недостаточности.
Рекомендуется всем пациентам исследование коагулограммы (ориентировочное исследование системы гемостаза) с определением протромбинового индекса, уровня фибриногена, протромбинового (тромбопластинового) времени в крови, активированного частичного тромбопластинового времени, тромбинового времени в крови, международного нормализованного отношения всем пациентам с целью исключения витамин-К-зависимой коагулопатии и оценки синтетической функции печени [39] .
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: витамин-К-зависимая коагулопатия может проявляться снижением протромбинового индекса (ПТИ) (удлинение протромбинового времени (ПВ)) и/или повышением международного нормализованного отношения (МНО), что связано с нарушением процессов всасывания витамина К в кишечнике. Данные изменения, наряду со снижением фибриногена, могут возникать при развитии печеночно-клеточной недостаточности.
Рекомендуется исследование уровня желчных кислот в крови всем пациентам с синдромом холестаза с целью оценки тяжести течения синдрома холестаза [13,29,36,37].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: исследование проводится при наличии соответствующей возможности в учреждении.
Рекомендуется провести комплекс исследований для диагностики нарушений функции щитовидной железы всем новорожденным с синдромом холестаза с целью выявления их дефицита [37,40].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 4)
Комментарии: комплексы могут включать в себя исследование уровня свободного трийодтиронина (СТ3) в крови, уровня свободного тироксина (СТ4) сыворотки крови, уровня тиреотропного гормона (ТТГ) в крови.
У новорожденных с гипогликемией в сочетании с синдромом холестаза рекомендуется комплексное определение концентрации тропных гормонов (исследование уровня тиреотропного гормона (ТТГ) в крови, исследование уровня адренокортикотропного гормона в крови, исследование уровня соматотропного гормона в крови) с целью исключения гипопитуитаризма [40–44].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 4)
Всем новорожденным с синдромом холестаза рекомендуется проведение исследования антител к потенциальным возбудителям гепатита методом иммуноферментного анализа (ИФА) (определение антител к цитомегаловирусу (Cytomegalovirus) в крови, антител к вирусу Коксаки (Coxsacki virus) в крови, антител к вирусу гепатиту С (Hepatitis С virus) в крови, антител к вирусу простого герпеса (Herpes simplex virus) в крови, антител к токсоплазме (Toxoplasma gondii) в крови, антител к бледной трепонеме (Treponema pallidum) в крови для определения этиологии гепатита [45–47] [48,49] [98,99].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Всем новорожденным с синдромом холестаза рекомендуется проведение ПЦР-исследование крови с целью выявления потенциальных возбудителей неонатального гепатита (определение ДНК вируса Эпштейна-Барр (Epstein - Barr virus) методом ПЦР в периферической и пуповинной крови, количественное исследование, определение ДНК цитомегаловируса (Cytomegalovirus) методом ПЦР в периферической и пуповинной крови, количественное исследование, РНК вируса гепатита С (Hepatitis С virus) в крови методом ПЦР, количественное исследование, антигена вируса гепатита С (Hepatitis С virus) в крови, ДНК вируса гепатита В (Hepatitis В virus) в крови методом ПЦР, количественное исследование, антигена (HbsAg) вируса гепатита В (Hepatitis В virus) в крови, РНК вируса краснухи (Rubella virus) методом ПЦР в периферической и пуповинной крови, качественное исследование, ДНК простого герпеса 1 и 2 типов (Herpes simplex virus types 1, 2) методом ПЦР в крови, количественное исследование, ДНК токсоплазмы (Toxoplasma gondii) методом ПЦР в периферической и пуповинной крови) [45–47] [48,49,98,99].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 4)
Рекомендовано определение аминокислот и ацилкарнитинов в сухом пятне крови методом тандемной масс-спектрометрии (комплексное определение концентрации на аминокислоты и ацилкарнитины в крови методом тандемной масс-спектрометрии) всем новорожденным с клиническими признаками холестаза с целью дифференциальной диагностики с другими наследственными нарушениями обмена веществ [50–53].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 4)
Комментарии: отсутствие патологических отклонений по данным неонатального скрининга не исключает наличие у ребенка метаболических нарушений и при прогрессирующем синдроме холестаза требует повторного проведения, в связи с вероятностью ложно-отрицательных результатов. Исследование проводится при наличии соответствующей возможности в учреждении.
Рекомендовано комплексное определение содержания органических кислот в моче методом газовой хроматографии c масс-спектрометрией (Комплексное определение содержания органических кислот в моче методом тандемной масс-спектрометрии) всем новорожденным с признаками синдрома холестаза с целью диагностики нарушения синтеза желчных кислот [54–57].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 4)
Комментарии: исследование проводится при наличии соответствующей возможности в учреждении. В спектр органических аминокислот в моче должно быть включено определение содержания желчных кислот.
Всем новорожденным с синдромом холестаза неясного генеза, с целью исключения наследственных заболеваний печени, рекомендовано проведение комплекса исследований для оценки холестатического синдрома (молекулярно-генетического исследования, определение патогенных вариантов в генах, ответственных за развитие наследственных заболеваний печени методом таргетного высокопроизводительного секвенирования) [21,59,61].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарий: при наличии признаков, высоко вероятно указывающих на какое-либо наследственное заболевание печени, рекомендуется таргетное исследование гена для выявления патогенного варианта методом секвенирования по Сенгеру. При отсутствии характерной клинической картины, целесообразно проведение расширенного молекулярно-генетического исследования методом высокопроизводительного секвенирования генов, ассоциированных с наследственными заболеваниями, сопровождающимися холестазом, или полноэкзомного секвенирования. Исследование проводится при наличии соответствующей возможности в учреждении.
Рекомендовано провести исследование кислотно-основного состояния и газов крови (исследование уровня водородных ионов (рН) в крови, уровня молочной кислоты в крови, уровня буферных веществ в крови) пациентам с синдромом холестаза [36,37,57,58].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Рекомендовано исследование уровня аммиака в крови новорожденным с синдромом холестаза, с диагностической целью и с целью своевременной коррекции терапии [36,37,58,62–66].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: исследование проводится при наличии соответствующей возможности в учреждении.
Рекомендовано исследование уровня жирорастворимых витаминов в крови (исследование уровня 1,25-ОН витамина Д в крови) новорожденным с синдромом холестаза, с целью выявления гиповитаминоза на фоне мальабсорбции жиров и своевременной коррекции терапии [36,37,58,62–66].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Рекомендуется проведение ультразвукового исследования органов брюшной полости (комплексного) пациентам с синдромом холестаза с целью выявления гепатоспленомегалии, аномалий развития желчевыводящей системы, признаков портальной гипертензии [67].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 3)
Комментарии: при транзиторном неонатальном холестазе при ультразвуковом исследовании (УЗИ) отмечают неспецифические изменения в виде различной степени выраженности увеличения размеров печени, слабого повышения эхогенности её паренхимы.
Характерными для БА изменениями являются: желчный пузырь натощак не визуализируется или выявляется в виде гиперэхогенного «тяжа»; имеет значение размер желчного пузыря (ЖП): его длина менее 1,5 см, а также наличие триады «GB-Ghhost»: длина менее 1,9 см, нерегулярный контур и отсутствие гладкой регулярной эхогенной слизистой оболочки ЖП; реакция на прием пищи отсутствует в течение всего промежутка между кормлениями; симптом «треугольного рубца» выявляется у большинства пациентов с БА, чувствительность, специфичность и точность в отношении БА для данного симптома составляют 85%, 100% и 95% соответственно. Это гиперэхогенный продолговатый тяж, локализующийся над стволом воротной вены. При наличии треугольного рубца и толщине фиброзной площадки более 3,4 мм вероятность БА увеличивается. До 1-1,5 месяцев «треугольный рубец» может быть не ярко выражен либо полностью отсутствовать у пациентов с БА в 30- 40%, а после 2-2,5 месяцев трудно отличим от перипортального фиброза, который может встречаться и при других заболеваниях печени, что затрудняет диагностику БА. Допплерографические маркеры БА: диаметр правой печеночной артерии более 1,5 мм и соотношение правой печеночной артерии и воротной вены более 0,45, средний диаметр печеночной артерии у пациентов с БА значительно больше, чем у пациентов с другими заболеваниями печени. К дополнительным критериям можно отнести расширение внутрипеченочных желчных протоков, реже - кисты в воротах печени, у 7-15 % детей с БА выявляется фрагментация селезенки с фиброзными тяжами в паренхиме, либо множественные добавочные дольки (полиспления).
УЗ-признаком кисты желчного протока является полостное образование в проекции общего желчного протока.
Однако при большинстве других наследственных заболеваниях печени характерные ультразвуковые признаки могут отсутствовать.
Рекомендуется проведение ультразвукового исследования почек всем пациентам с целью выявления особенностей строения почек [33,68–70].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 4)
Комментарии: у пациентов с синдромом холестаза особенности развития почек могут являться одним из внепеченочных признаков ряда наследственных заболеваний печени, таких как синдром Алажилля, при котором отмечается повышение эхогенности коркового слоя паренхимы почек, снижение дифференцировки почечной ткани на корковое и мозговое вещество за счет уменьшения количества пирамидок в срезе до 4 и менее, а также имеются особенности строения пирамидок в виде вытянутой формы и различные кистозные изменения в пирамидках и паренхиме [71].
Рекомендуется проведение эхокардиографии всем пациентам с синдромом холестаза для исключения аномалий развития сердечно-сосудистой системы [72].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4)
Комментарии: врожденные пороки сердца могут являться одним из внепеченочных признаков ряда наследственных заболеваний печени, в частности сужение легочной артерии и ее ветвей, являются одним из главных клинических признаков синдрома Алажилля.
Рекомендуется проведение рентгенографии грудного отдела позвоночника всем пациентам с подозрением на синдром холестаза с целью выявления характерных аномалий развития позвонков [37,73].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: аномалии позвонков, в частности «бабочковидная» деформация позвонков наблюдаются у пациентов с синдромом Алажилля.
Рекомендуется прием (осмотр, консультация) врача-офтальмолога первичный всем пациентам с подозрением на синдром Алажилля с целью исключения эмбриотоксона [18,74].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: задний эмбриотоксон может быть выявлен у 78-89% пациентов с СА.
Всем пациентам с неонатальным холестазом при подозрении на билиарную атрезию рекомендуется прием (осмотр, консультация) врача-детского хирурга первичный с целью определения показаний к хирургическому лечению [3,75,76].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: при подозрении на билиарную атрезию, рекомендуется консультация врача-детского хирурга с целью решения вопроса о проведении порто-энтеростомии по Касаи.
Всем пациентам с неонатальным холестазом неясной этиологии рекомендуется прием (осмотр, консультация) врача-генетика первичный с целью определения объема молекулярно-генетических методов диагностики [77].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
3.1 Консервативное лечение
3.1.1. Лечебное питание
Без адекватного оттока желчи желчные кислоты не могут нормально секретироваться в просвет кишечника. Соли желчных кислот являются необходимым компонентом мицеллообразования в просвете тонкой кишки. Мицеллы позволяют поглощать нерастворимые молекулы, включая большинство липидов и жирорастворимых витаминов. При синдроме холестаза секреция желчных солей снижена, что приводит к неэффективному всасыванию жира. Нарушение всасывания из-за холестаза может привести к задержке роста, потере веса и дефициту жирорастворимых витаминов, включая витамин А, витамин D, витамин Е и витамин К [78–81].
Новорожденным с синдромом холестаза рекомендуется питание специализированным продуктом детского диетического (лечебного) с повышенным содержанием среднецепочечных триглицеридов с целью предотвращения нарушений липидного обмена и дефицита жирорастворимых витаминов [82,83].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: в условиях мальабсорбции жиров при синдроме холестаза важным компонентом лечебного питания являются среднецепочечные триглицериды. Среднецепочечные триглицериды (СЦТ) обладают большей водорастворимостью, абсорбируются в желудке и тонкой кишке без участия желчных кислот. Оптимальная доля общих липидов в качестве СЦТ составляет от 30 до 50%. Следует избегать более высокого содержания СЦТ в рационе (>80%) без адекватного добавления полиненасыщенных жирных кислот (ПНЖК), т.к. это может привести к дефициту незаменимых жирных кислот (НЖК). ПНЖК также являются предшественниками эйкозаноидов, которые улучшают иммунную функцию, уменьшают системное воспаление и участвуют в агрегации тромбоцитов. Дефицит ПНЖК и олигоненасыщенных жирных кислот (ОНЖК) может быть результатом низкого потребления, нарушения переваривания/мальабсорбции жиров и неэффективного удлинения предшественников ОНЖК. Дефицит также может быть ятрогенным, если в рационе очень много СЦТ и мало ДЦТ.
Развитие холестаза, обусловленного внепечёночной перинатальной патологией, чаще наблюдают у недоношенных, и в том числе глубоко недоношенных детей, в связи с чем при выборе лечебного питания необходимо учитывать следующие составляющие:
степень выраженности и длительность холестаза;
гестационный и постнатальный возраст ребёнка;
нарушение расщепления и всасывания других компонентов, в том числе белков и углеводов.
У детей с тяжёлой перинатальной патологией можно наблюдать нарушение процессов всасывания белков и углеводов, что наряду с синдромом мальабсорбции жиров, вследствие холестаза, служит показанием для использования соответствующего лечебного питания. В данном случае целесообразно применять смесь на основе белкового гидролизата, не содержащую лактозу, в состав которой входит 50% среднецепочечных триглицеридов. У недоношенных детей, и особенно глубоко недоношенных показано назначение смесей, предназначенных для вскармливания недоношенных детей (пре формулы), обогащенных среднецепочечными триглицеридами. Следует отметить отсутствие противопоказаний к использованию грудного молока. Вместе с тем только грудное вскармливание не может обеспечить потребность ребёнка в основных ингредиентах и, прежде всего, в жировом компоненте, в связи с чем целесообразно комбинировать грудное молоко с лечебной смесью под контролем динамики веса.
Рекомендуется использование молочной смеси - специализированного продукта диетического лечебного питания для детей с рождения и взрослых при дефектах окисления длинноцепочных жирных кислот, хилотораксе и лимфангиэктазии "Monogen" ("Моноген") (при нарушении обмена жирных кислот)**** в объеме 1/3-1/2 суточного рациона.
3.1.2. Лекарственная терапия
У новорожденных с транзиторным холестазом общими подходами к терапии являются: адекватное лечение основного заболевания, исключение или ограничение потенциально гепатотоксичных лекарств и препаратов крови, раннее начало энтерального питания, а также желчегонная терапия. При выборе желчегонной терапии необходимо учитывать морфофункциональные особенности гепатобилиарной системы у новорождённых, включающие высокий уровень синтеза первичных жёлчных кислот и незрелость экскреторных механизмов.
Рекомендуется назначение #урсодезоксихолевой кислоты** (код АТХ A05AA02) в дозе 20-30 мг/кг/сутки в два приема перорально длительно всем новорожденным с синдромом холестаза с желчегонной целью [84].
Уровень убедительности рекомендаций А (уровень достоверности доказательств – 1)
Комментарии: Урсодезоксихолевая кислота** (код АТХ A05AA02) представляет собой третичную желчную кислоту. Его предполагаемые механизмы действия включают усиление экскреции желчи и изменение состава желчных кислот до более гидрофильных и менее токсичных.
Рекомендуется назначение витаминов - #ретинол** (витамин А) (код АТХ A11CA), колекальциферол** (код АТХ A11CC), витамин Е (код АТХ A11HA), фитоменадион (код АТХ B02BA) пациентам с синдромом холестаза с целью восполнения их дефицита [80].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4)
Комментарии: дефицит жирорастворимых витаминов из-за стеатореи часто встречается при хроническом холестазе. К ним относятся витамины А, D, Е и К. Большинству пациентов с синдромом холестаза требуется добавление одного или более из этих жирорастворимых витаминов. Дефицит витамина Е может привести к неврологическим симптомам, включая периферическую нейропатию, мозжечковую атаксию, дегенерацию периферических аксонов и задних столбовых нейронов. Дефицит витамина А может привести к глазным заболеваниям, включая гемералопию, высыхание или изъязвление роговицы, ретинопатию и, в конечном итоге, к слепоте. Описаны ксерофтальмии и кератомаляции вследствие дефицита витамина А у ребенка с СА. Витамин К необходим для синтеза нескольких факторов свертывания крови, включая факторы II, VII, IX и X. Препараты группы Витамин К (код АТХ B02BA) обычно назначаются перорально, хотя пациентам с тяжелым холестазом может потребоваться парентеральное введение. Одновременное введение водорастворимого витамина Е (код АТХ A11HA) с витамином D и его аналогами (код АТХ A11CC) может увеличивать всасывание витамина D [79,85].
Жирорастворимые витамины назначаются при длительности холестаза более 10 дней, в следующих дозировках:
Колекальциферол**(код АТХ A11CC) перорально 1000-2500 МЕ/сутки длительностью не менее 1 месяца. Коррекция дозы проводится под контролем исследования уровня 1,25-OH витамина Д в крови.
#Ретинол** (код АТХ A11CA) перорально 3000-10000 МЕ/сутки [4].
Витамин Е (код АТХ A11HA) перорально 50 мг/сутки, если масса тела больше 1 кг, 25 мг/сутки если масса тела меньше 1 кг.
Группа Витамин K (код АТХ В02ВА) [4,12,86–93]:
#Фитоменадион (код АТХ В02ВА01) 1% эмульсия. Режим назначения профилактической дозы варьирует от 1 мг/кг раз в неделю у детей массой тела менее 5 кг до 2,5 – 5,0 мг от 1 до 3 раз в неделю или 10 мг 1 раз в 2 недели у детей массой тела более 5 кг. Доза и кратность профилактики подбирается индивидуально в зависимости от результатов коагулограммы и в тяжелых случаях может доходить до ежедневного приема. При наличии холестаза или синдрома мальабсорбции целесообразно парентеральное введение препаратов группы витамина К (код АТХ В02ВА). При отсутствии #фитоменадиона (код АТХ В02ВА01) допустимо использовать #менадиона натрия бисульфит** (код АТХ B02BA02) 1% раствор. Необходимо учитывать, что его действие начнется через 8-24 часа после введения.
3.2 Хирургическое лечение
Хирургическое лечение синдрома холестаза в неонатальном периоде не проводится за исключением случаев, когда высоко вероятен диагноз Билиарная атрезия, при которой проводится операция печеночной гепатопортоэнтеростомии по Касаи. В настоящее время используется минилапаротомный доступ или лапароскопическое вмешательство.
Трансплантация печени в неонатальном периоде не проводится.
Лечение кисты общего жёлчного протока только хирургическое – полное иссечение кисты с формированием билиодигестивного анастомоза c изолированной петлёй тощей кишки по Ру [30].
3.3 Иное лечение
Не применимо.
Проводится профильными специалистами (врач-педиатр, врач-гастроэнтеролог, врач-офтальмолог, врач-детский хирург и др.) в зависимости от сопутствующей патологии и осложнений.
Детям с синдромом холестаза вследствие наследственных заболеваний печени рекомендуется диспансерный прием (осмотр, консультация) врача-педиатра с целью профилактики осложнений заболевания [1,36].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: после выписки из стационара рекомендуется проведение диспансерного наблюдения: физикальный осмотр специалиста (врача-педиатра, врача-гастроэнтеролога, врача-детского хирурга и др.), который наблюдает пациента, оценивая проявления заболевания 1 раз в 1-3 месяца в течение первого года жизни, далее 1 р в 3-6 месяцев на втором году жизни. При каждом посещении специалиста осуществляется контроль активности заболевания: общий (клинический) анализ крови и общий (клинический) анализ мочи (протеинурия), биохимический общетерапевтический анализ крови (исследование уровня общего белка в крови, исследование уровня альбумина в крови, уровня общего билирубина в крови, уровня билирубина свободного (неконъюгированного) в крови, уровня билирубина связанного (конъюгированного) в крови, уровня триглицеридов в крови, уровня холестерина в крови; определение активности креатинкиназы в крови, активности аспартатаминотрансферазы в крови, активности аланинаминотрансферазы в крови, активности гамма-глютамилтрансферазы в крови, активности щелочной фосфатазы в крови), ориентировочное исследование системы гемостаза с определением протромбинового индекса, уровня фибриногена, протромбинового (тромбопластинового) времени в крови, активированного частичного тромбопластинового времени, тромбинового времени в крови, международного нормализованного отношения (МНО).
Детям с синдромом холестаза, вследствие наследственных заболеваний печени рекомендуется прием (осмотр, консультация) врача-генетика повторный с целью медико-генетического консультирования [77,94].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 5)
Комментарии: рекомендуется прием врача-генетика родителям после установления диагноза ребенку с целью медико-генетического консультирования по вопросу генетического риска рождения в этой семье детей с наследственной патологией.
Детям с синдромом холестаза рекомендуется вакцинация с целью профилактики инфекционных заболеваний [95].
Уровень убедительности рекомендаций C (уровень достоверности доказательств – 5)
Комментарии: рекомендуется вакцинировать новорожденных с неонатальным холестазом в соответствии с национальным календарем прививок при отсутствии противопоказаний со стороны других органов и систем. Для профилактики вирусного гепатита В всем новорожденным в течение первых 24 ч жизни вводят вакцину для профилактики вирусного гепатита В** (код АТХ J07BC01, затем в возрасте 1 и 6 месяцев. Если мать – носитель HBSAg, то проводят дополнительную вакцинацию в возрасте 12 месяцев.
Показания для госпитализации в медицинскую организацию:
1) Тяжелое нестабильное состояние ребенка.
2) Необходимость коррекции гипоальбуминемии.
Показания к выписке пациента из медицинской организации
1) Стабильное удовлетворительное состояние ребенка.
2) Стабилизация лабораторных показателей, отсутствие гипоальбуминемии.
3) Отсутствие противопоказаний к выписке.
В случае подозрения на порок развития желчевыводящих путей, пациент направляется в профильное хирургическое отделение для проведения дополнительных методов исследования и хирургического лечения в условиях стационара.
Исходы синдрома холестаза зависят от этиологии заболевания.
